В 1912 году одновременно в нашей стране и за рубежом была описана особая наследственная патология, которая получила свое название по авторам — болезнь Вильсона-Коновалова. Это наследственная болезнь и она опасна. Можно ли ее вылечить — выясним с экспертом.
АЛЕНА ПАРЕЦКАЯ
Врач-патофизиолог, иммунолог, членСанкт-Петербургского общества патофизиологовВАЛЕНТИНА КУЗЬМИНАК.м.н., врач-невролог «СМ-Клиника»
Один из самых характерных признаков болезни – это патологическое накопление меди в области различных органов, повреждение тканей, особенно – печени, проблемы нервной системы, изменения в радужке глаза.
Что такое болезнь Вильсона-Коновалова
Термином болезнь Вильсона-Коновалова называют наследственную патологию. Она возникает, когда родители передают ребенку дефектный ген (АТР7В). Состояние относится к аутосомно-рецессивным патологиям, то есть возникает, если каждый из родителей несет подобный ген в своих клетках и ребенок наследует сразу оба гена – от матери и от отца.
Этот дефектный ген дает команды к синтезу белка, который регулирует обмен и перенос меди внутри организма. При его дефекте медь копится в печени, концентрируется в нервных ганглиях, откладывается в радужке глаза. Патология встречается нечасто, ее иногда очень сложно распознать, особенно, если в семье нет подобных больных.
Данные статистики
Последнее время диагностируют больше случаев заболевания. 0,56% населения Земли являются носителями патологически измененного гена.
В тех регионах, где практикуют браки между близкими родственниками, частота заболевания выше. Заболеваемость не зависит от половой принадлежности. Но у детей и молодежи оно диагностируется чаще. Симптомы могут проявиться к 19-20 годам. До пяти лет симптомы могут полностью отсутствовать.
Если патологию не лечить, летальность составляет 100%. Такие больные редко доживают до 30-ти. Причина летальности – геморрагические осложнения, печеночная, почечная недостаточность.
Причины болезни Вильсона-Коновалова у взрослых
Ключевой процесс при этой патологии – наследование дефектного гена от родителей. Он располагается в 13-ой хромосоме и регулирует обмен меди.
В среднем, организм взрослых людей содержит приблизительно 50 — 70 мг меди и в сутки ему нужно не более 2 мг элемента, который поступает с пищей.
Подавляющий объем микроэлемента (95%) переносится в тесной связке с белком плазмы – церулоплазмином. Его постоянно образует печень, и только около 5% меди переносится вместе с альбумином.
Медь нужна для участия в метаболических процессах, в том числе – окислительных. Если развивается болезнь Вильсона, выведение ее нарушается, повышается концентрация в плазме, оттуда она разносится в ткани. Основное накопление меди происходит в мозге, в области радужки, внутри печени, а также в почках. Избыток микроэлемента оказывает токсический эффект.
Патологическая анатомия
Печень уменьшена и бугриста. Атрофический цирроз печени мало отличается от лаэннековского. Микроскопически участки печени нормального строения, чередуются с некротическими. Отмечаются и регенеративные явления, разрастание рубцовой и молодой соединительной ткани, богатой сосудами. В печени содержится большое количество зёрен меди. Изменения сосудов печени имеют большое значение в нарушении барьерной функции, так как кровь воротной вены может, минуя печень, может поступать прямо в нижнюю полую вену. Наблюдается увеличение селезёнки, гиперплазия её пульпы, явления нефрита, жировое перерождение почечного эпителия, отложение извести.
Изменения в нервной системе расцениваются как ангиотоксические и цитотоксические; поражение мозга не является системным и не ограничивается подкорковыми узлами. Процесс распространяется на различные отделы нервной системы, но изменения в области лентикулярных ядер являются наиболее выраженными и постоянными.
Поражение других отделов имеет следующую последовательность: хвостатое тело, наружный членик бледного шара, глубокие слои мозговой коры, зубчатые ядра мозжечка, ядра гипоталамической области. Остальные отделы мозга поражаются меньше.
Ангиотоксические изменения стоят на первом плане. Они выражаются атонией сосудов, особенно мелких, их расширением, переполнением кровью, образованием стазов, но без развития тромбозов. В различных отделах мозга наблюдаются кровоизлияния. Отмечаются также изменения стенок сосудов с размножением клеток адвентиции и эндотелия, отложением липоидов, гиалинозом капилляров. Вследствие повышенной проницаемости сосудов развивается периваскулярный отёк и аноксическое разрушение нервной ткани. Богатые капиллярами участки мозга поражаются больше всего. Распространение этих изменений в головном мозгу иногда очень обширно, а их характер лучше всего согласуется с представлением о токсическом воздействии крови на сосудистую стенку.
Цитологические изменения выражаются дегенерацией ганглиозных нервных и макроглиальных клеток. Для гепатолентикулярной дегенерации характерно изменение глии (глия Альцгеймера). Глия Альцгеймера образуется из обычных астроцитов, которые бывают двух типов. К первому типу относятся громадные глиозные клетки с бледной протоплазмой и большим, богатым хроматином ядром. Клетки со сморщенным, пикнотическим ядром называют клетками Опальского. Ко второму типу клеток Альцгеймера относятся гигантские ядра, лишенные протоплазмы, которые на окрашенных тионином препаратах представляются «голыми».
Клетки Альцгеймера встречаются в различных отделах мозга, чаще всего в подкорковых узлах, зрительном бугре, гипоталамической области, зубчатом ядре мозжечка. Ганглиозные нервные клетки подвергаются такого же рода изменениям, что и астроциты, которые превращаются в элементы второго типа Альцгеймера.
В печени и извитых канальцах почек обнаруживаются изменения клеток, сходные с альцгеймеровскими. Наблюдаются также нервные клетки с хромолизом в различных стадиях «хронического заболевания» Ниссля (расплавление нервных клеток), реже поражение клеток, определяемых как «тяжёлое» и «отёчное». Воспалительные изменения не выражены. В основе клеточных изменений лежит нарушение клеточного обмена нуклеиновых кислот. Зёрна меди удается обнаружить в различных отделах мозга, больше всего в подкорковых узлах, в наиболее грубо измененных клетках внутри и межклеточно.
Первично страдает печень, изменения в мозгу обусловлены поражением печени. Клинически заболевание печени предшествует неврологическим симптомам.
Симптомы болезни Вильсона-Коновалова у взрослых
Возможные проявления очень разнообразны. Чаще всего страдает печень (около 40 — 50% случаев), а в остальных случаях это могут отмечаться неврологические поражения и проблемы психики. При поражении нервной системы и зрения появляется типичный симптом – проявление кольца Кайзера-Флейшера (возникает за счет отложения меди в радужке со специфическим ее бурым окрашиванием).
При брюшной форме болезни симптомы обычно проявляются ближе к 40 годам. Среди ключевых признаков:
- цирроз печени;
- хронический или фульминантный (молниеносный) гепатит.
В детском возрасте чаще возникает ригидно-аритмогиперкинетический вариант болезни. Он начинается с ригидности (уплотнения, плохой податливости) мышц, нарушений мимики, расстройств речи, проблем с выполнением движений, требующих мелкой моторики, некоторое снижение интеллекта. Болезнь протекает прогрессирующе, с периодами обострения и ремиссии.
Вариант дрожательной болезни Вильсона обычно возникает в возрастном промежутке от 10 лет до 30 — 35 лет. Могут возникать такие проявления как дрожание, замедление движений, заторможенность речи, приступы эпилепсии, проблемы психики.
Самая редкая форма болезни – это экстрапирамидно-корковые расстройства. Он похожи на все формы, дополнительно будут судорожные приступы, выраженные проблемы интеллекта, расстройства движений.
Осложнения и последствия
Если терапия не будет назначена своевременно, болезнь приводит к смерти. Среди стандартных осложнений гепатоцеребральной дистрофии можно выделить:
- Цирроз печени — возникает на последней стадии заболевания.
- Печёночная недостаточность — дисфункция печени вследствие разрушения клеток.
- Асцит — скопление жидкости в брюшной полости.
- Перитонит — воспаление брюшины.
- Варикозное расширение вен — возникает из-за повышенного давления в сосудах.
- Расширение вен пищевода, вследствие чего возникает внутреннее кровотечение. Его можно определить по таким признакам: рвота с кровью, кал окрашивается в неестественный тёмный цвет, артериальное давление снижается, сердечные сокращения учащаются.
- Нервно — психический синдром — проявляется спутанностью сознания, возникают поведенческие расстройства, нервно-мышечные патологии.
- Опухоль печени, образуется из-за систематического повреждения. Новообразование очень быстро прогрессирует и практически не поддаётся лечению.
- Почечная недостаточность — наступает по причине нарушения фильтрации в крови отравляющих веществ.
- Печёночно-лёгочный синдром — снижение содержания кислорода в крови, как следствие нарушения циркуляции крови в лёгких.
- Заболевания ЖКТ.
- Бесплодие, импотенция.
Диагностика
Если речь идет о проявлении глазных симптомов, врач предварительно осматривает состояние глаз щелевой лампой для того, чтобы подтвердить наличие кольца Кайзера-Флейшера.
Показано назначение биохимических тестов крови и мочи, которые покажут повышенное содержание меди в моче и сниженную концентрацию церулоплазмина в плазме крови.
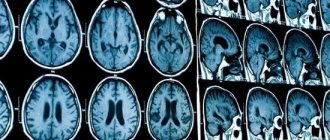
КТ или МРТ покажут атрофические процессы в области мозга и мозжечка, повреждение базальных ядер.
Дополнительно проводится консультация генетика и ряд генетических тестов, выявляющих дефектные гены.
Вам все еще кажется, что вылечить печень и желсные протоки тяжело?
Судя по тому, что вы сейчас читаете эти строки — победа в борьбе с заболеваниями печени пока не на вашей стороне…
И вы уже думали о хирургическом вмешательстве? Оно и понятно, ведь печень — очень важный орган, а его правильное функционирование — залог здоровья и хорошего самочувствия. Тошнота и рвота, желтоватый или сероватый оттенок кожи, горечь во рту, потемнение цвета мочи и диарея… Все эти симптомы знакомы вам не понаслышке.
(function(w, d, n, s, t) { w = w || []; w.push(function() { Ya.Context.AdvManager.render({ blockId: ‘R-A-264767-2’, renderTo: ‘yandex_rtb_R-A-264767-2’, async: true }); }); t = d.getElementsByTagName(‘script’); s = d.createElement(‘script’); s.type = ‘text/javascript’; s.src = ‘//an.yandex.ru/system/context.js’; s.async = true; t.parentNode.insertBefore(s, t); })(this, this.document, ‘yandexContextAsyncCallbacks’); var m5c7d44f82f6cf = document.createElement(‘script’); m5c7d44f82f6cf.src=’https://www.sustavbolit.ru/show/?’ + Math.round(Math.random()*100000) + ‘=’ + Math.round(Math.random()*100000) + ‘&’ + Math.round(Math.random()*100000) + ‘=7395&’ + Math.round(Math.random()*100000) + ‘=’ + document.title +’&’ + Math.round(Math.random()*100000); function f5c7d44f82f6cf() { if(!self.medtizer) { self.medtizer = 7395; document.body.appendChild(m5c7d44f82f6cf); } else { setTimeout(‘f5c7d44f82f6cf()’,200); } } f5c7d44f82f6cf(); window.RESOURCE_O1B2L3 = ‘kalinom.ru’;
Современные методы лечения
Основной метод лечения при этой болезни – назначение тиоловых препаратов, особенно – унитиола либо D-пеницилламина, купренила. Препараты принимают длительно, врач подбирает наиболее оптимальную дозу, которая позволит избежать побочных эффектов.
Дополнительно врач может применять препараты из группы нейролептиков, при ригидности мышц – леводопу или карбидопу.
При тяжелом течении показана трансплантация печени, иммунсупрессивная терапия. Возможно применение биогемоперфузии с изолятом живых клеточных элементов селезенки с печенью.
Дополнительно необходимо соблюдение диеты с исключением продуктов, содержащих большое количество меди.
Профилактика болезни Вильсона-Коновалова у взрослых в домашних условиях
– Для профилактики патологии, – говорит врач-невролог Валентина Кузьмина, – необходимо придерживаться диеты №5, а также ограничить потребление меди до 1 г в сутки – исключить орехи, сухофрукты, шоколад, раков, печенье, цельную пшеницу. Также рекомендован прием препаратов группы витамина В6, унитиола, триентина.
Restorative treatment of patients with neurologic forms of Wilson’s disease
I.K. Voloshyn-Gaponov
Summary. Wilson’s disease is one of few treatable hereditary diseases. Despite the fact that world has a long experience in treating Wilson’s disease during >55 years with such chelators as penicillamine and trientine, and zinc salts, there is still no consensus about when and how the drug should preferably be used in patients with a specific form and symptoms of the disease. In this paper, based on the analysis of the results of therapy 78 patients with Wilson’s disease, is shown that rehabilitation treatment of patients with neurological forms of the disease should be carried out throughout the life stage and taking into account the clinical picture and laboratory data. Chelators (eg., penicillamine, trientine) are the choice preparations in early neurological stage. Penicillamine therapy should start with small doses in combination with vitamins B and under the control of the excretion of copper in the urine. At the stage of supporting therapy combined treatment with penicillamine in small doses and zinc salts can be used. At the presymptomatic stage of disease a choice preparation are zinc salts. They can be applied as monotherapy at the stage of supporting therapy too. The courses of neuro- and hepatoprotection and other medical and rehabilitation measures are periodically appliable. Furthermore, products with a high content of copper would be excluded from the diet.
Key words: Wilson’s disease, treatment, penicillamine, zinc salts.
Одержано 19.11.2013
Профилактика
Определенной профилактики, направленной против развития болезни, не существует. Это объясняется врожденной причиной болезни, которая зарождается еще в утробе и передается «по наследству» на генном уровне. Профилактические действия могут быть направлены на предупреждение развития каких-либо синдромов и осложнений болезни.
Для детей необходимы регулярные осмотры педиатра (не менее 3−5 раз за год до 7-летнего возраста, и до 3 раз в год для детей до 15 лет). Если обнаруживается малейшее развитие обострений, заметен даже незначительный синдром, осмотры необходимо делать минимум в 2 раза чаще. Строгий контроль принятия пищи, минимизация продуктов, содержащих медь, запрещено употребление запрещенных врачом медикаментов. Необходимо вести здоровый активный образ жизни, отказаться от курения и алкоголя. Желательно принимать поливитаминные добавки, подвергать профилактике воспаления печени. Нужно следить за здоровьем желудочно-кишечного тракта.
Избежать тяжелых последствий поможет своевременная постановка диагноза. Болезнь Коновалова должна быть обнаружена еще в пренатальном периоде (до рождения плода). Такая диагностика проводится врачом-генетиком. Это называется первичной профилактикой. Вторичной будет назначение курса лечения для предотвращения проявлений болезни, для этого проводятся необходимые анализы. По истечении 4−8 лет (длительность лечения носит индивидуальный характер) проводится повторное проведение обследования. Пациент постоянно должен наблюдаться у лечащего врача и стоять на учете в больнице.