Введение
В сфере детской практической гепатологии особое место занимают орфанные заболевания, протекающие с поражением печени, к которым, в частности, относится гликогеновая болезнь (ГБ). В нашей стране опыт курации таких пациентов ввиду редкости этой нозологии относительно невелик, а соответственно, отсутствует достаточное число врачей-специалистов в данной области. Клинико-лабораторная картина заболевания чрезвычайно многообразна, что часто затрудняет своевременную его диагностику, приводит к развитию тяжелых осложнений и инвалидизации больных. В связи с этим представляется актуальным изложить в настоящей статье современные сведения по этиологии, патогенезу, клинической картине, методам диагностики и лечениия ГБ у детей.
Классификация
В структуру понятия «гликогеновая болезнь» входит не менее 15 типов генетически обусловленных патологических состояний, в основе которых лежит нарушение синтеза или расщепления гликогена с накоплением последнего в различных органах и тканях, преимущественно в печени и мышцах. В настоящее время принято использовать классификацию, предложенную G. Cori в 1954 г. и построенную по хронологическому принципу: типы ГБ обозначаются римскими цифрами и располагаются в порядке открытия фенотипа и соответствующих ферментных дефектов [1]. Однако классификация постоянно пересматривается, что вызывает сложности при проведении дифференциальной диагностики между разными типами заболевания. Например, сейчас отсутствует деление на подтипы VIa и VIb. Также исключен из номенклатуры VIII тип ГБ. При этом значительно расширена классификация типа IX, в структуре которого выделено 5 подтипов (а1, а2, b, c, d). Кроме того, в классификацию добавлены новые типы – XIV, XV. Ранее индекс Х был присвоен заболеванию, связанному с дефицитом циклической 3,5-АМФ-зависимой киназы (описан у единственной больной), но впоследствии эта форма патологии была отнесена к типу VI. Сейчас к подтипу X относят заболевание, вызванное недостаточностью мышечной фосфоглицератмутазы-2. Хотя дефицит гликогенсинтазы не приводит к избыточному накоплению гликогена в печени, а наоборот – к его крайне низкому содержанию, эта форма патологии часто классифицируется как ГБ типа 0, поскольку она также является дефектом метаболизма гликогена и вызывает изменения, сходные с другими формами ГБ. Важно отметить, что у одного пациента возможно сочетание нескольких ферментных дефектов, и в таких случаях принято говорить о неидентифицированных типах этого заболевания [2]. В настоящей статье мы коснемся только тех типов ГБ, которые протекают с преимущественным поражением печени (типы I, III, VI и IX).
Этиология
Гликогеновую болезнь Iа-типа вызывают мутации гена G6PC, кодирующего глюкозо-6-фосфатазу, что приводит к ее недостаточности в печени, почках, слизистой оболочке кишечника [3, 4], а также, по некоторым данным, – в островках β-клеток поджелудочной железы и желчного пузыря [5]. Причиной возникновения ГБ Ib-типа являются мутации гена SLC17A4, кодирующего микросомальный транспортный белок Т1 (транслоказу глюкозо-6-фосфатазы), что приводит к ее дефициту в печени, почках, слизистой оболочке кишечника [6]. Гликогеновая болезнь III типа возникает в результате мутаций гена AGL, кодирующего гликоген-деветвящий фермент, который имеет две каталитические единицы: амило-1,6-глюкозидазу и 4-альфа-глюканотрансферазу, способные функционировать независимо друг от друга, однако для нормального действия деветвящего фермента необходима активность обеих каталитических единиц. Подавляющее большинство больных имеют дефицит этого энзима как в печени, так и в мышцах (подтип IIIa), однако примерно у 15 % пациентов он отмечается только в печени (подтип IIIb). Наличие указанных подтипов объясняется различной экспрессией фермента в тканях [7–10].
Причиной возникновения ГБ VI- типа являются мутации гена PYGL, кодирующего фосфорилазу печени [11, 12]. Гликогеновая болезнь IX-типа развивается на фоне мутаций в гене PHKA2 – для IXa1, IXa2 подтипов, в PHKAB – для IXb подтипа [13, 14].
Патогенез
Описание основных звеньев патогенеза и клинико-лабораторных проявлений ГБ I типа может служить основанием для понимания симптомов всех остальных печеночных форм этой патологии. Причиной развития I типа ГБ является наследственный дефект глюкозо-6-фосфатазы – фермента, обеспечивающего выход глюкозы в кровоток после ее высвобождения из гликогена клеток печени. Таким образом, нарушается реакция образования свободной глюкозы из глюкозо-6-фосфата, следствием чего является гипогликемия. Кроме того, вследствие дефекта глюкозо-6-фосфатазы происходит накопление в клетках печени субстрата блокированной реакции – глюкозо-6-фосфата, который, вовлекаясь в процесс катаболизма, превращается в пируват и лактат. При этом в крови значительно повышается уровень лактата, поэтому возможно развитие метаболического лактат-ацидоза. В тяжелых случаях результатом гипогликемии могут быть судороги, кома и летальный исход. Гипогликемия сопровождается уменьшением содержания инсулина и снижением отношения инсулин/глюкагон, что в свою очередь ведет к ускорению липолиза жировой ткани (в результате действия глюкагона) и выходу в кровь жирных кислот. В крови повышается концентрация триглицеридов как результат снижения активности липопротеин липазы жировой ткани – фермента, активируемого инсулином и обеспечивающего усвоение триглицеридов клетками жировой ткани. Одновременно на фоне значительно возрастающего содержания глюкозо-6-фосфата в клетках увеличивается его использование в пентозофосфатном пути с повышенным образованием рибозо-5-фосфата – субстрата для синтеза пуриновых нуклеотидов, конечным продуктом которого является мочевая кислота. Снижается и выведение мочевой кислоты с образованием уратов вследствие увеличения продукции лактата и изменения рН мочи в кислую сторону, что затрудняет выведение уратов – труднорастворимых солей мочевой кислоты [2, 15–18].
Клиническая картина, лабораторные проявления
Дебют ГБ I типа происходит в первые дни после рождения ребенка. Главным признаком болезни является выраженная гипогликемия, возникающая даже при малейшем голодании. Ее клиническими проявлениями могут быть немотивированный продолжительный плач и беспокойство ребенка, повышенная потливость, бледность кожных покровов, цианоз носогубного треугольника, вялость; могут развиваться судороги. Вскоре присоединяется выраженная гепатомегалия, приводящая к значительному увеличению размеров живота, что изначально может ошибочно приниматься за вздутие или асцит и приводит к неверной интерпретации патологии. У детей с ГБ I типа могут отмечаться локальные отложения подкожной жировой клетчатки, преимущественно на щеках («кукольное» лицо), груди, ягодицах, бедрах [3, 18]. С возрастом заболевание прогрессирует, развиваются множественные осложнения. На фоне чрезмерного отложения гликогена в печени и выраженных дистрофических изменений гепатоцитов формируется фиброз с возможностью последующей цирротической трансформации. Также могут возникать аденомы печени, имеющие потенциальный риск малигнизации [19–21]. На фоне гиперлипидемии нередко развивается хронический панкреатит [22, 23]. В постпубертатный период на первый план выступает гиперурикемия с ее клиническими осложнениями в виде подагрического артрита. Почечные поражения при ГБ I типа проявляются фокально-сегментарным гломерулосклерозом, сопровождающимся протеинурией со снижением почечных функций, тубулярными нарушениями проксимального типа, напоминающими синдром ДеТони–Дебре–Фанкони, нефролитиазом и нефрокальцинозом [3, 5, 24]. Пациенты отстают в росте, отмечаются остеопороз, повышенная склонность к переломам. Может иметь место интермиттирующая диарея, напоминающая таковую при воспалительных заболеваниях кишечника (кроно-подобный синдром) [25].
У детей с ГБ Ib-типа ввиду выраженной нейтропении (вплоть до агранулоцитоза) отмечаются частые рецидивирующие инфекции с развитием гнойно-септических очагов [26]. Прогноз при ГБ I типа относительно благоприятный только при ранней диагностике и своевременно начатом лечении. Без специальной диеты, частых кормлений и самоконтроля прогноз неблагоприятный – большинство больных умирают в раннем детском возрасте от гипогликемической комы, присоединяющихся тяжелых инфекций, в старшем возрасте – от гепатоцеллюлярной карциномы и подагрической нефропатии [3, 18].
При ГБ III типа нарушается дегенерация гликогена, что ведет к образованию патологического гликогена, накапливающегося в гепатоцитах и мышцах и имеющего токсический эффект. Клинические проявления ГБ III типа во многом сходны с таковыми при I типе: гепатомегалия (при циррозе присоединяется спленомегалия), отставание в росте. Кроме того, отмечается кардиомегалия, мышечная слабость. Лабораторно имеют место гипогликемия и гиперлипидемия, умеренный ацидоз и кетонурия, синдром цитолиза. Прогноз в большинстве случаев благоприятный, однако описаны случаи прогрессирования заболевания с развитием цирроза печени и тяжелой миопатии [3, 18].
Для ГБ VI типа характерна гепатомегалия, гипогликемия, гиперхолестеринемия, повышение уровней трансаминаз в сыворотке крови. Прогноз благоприятный, т.к. нарушения в обмене гликогена компенсируются за счет глюконеогенеза. Проявления ГБ IX практически идентичны таковым при VI типе [18].
Диагностика
Как уже говорилось, ГБ можно заподозрить на основании совокупности описанных клинико-лабораторных признаков. Однако для окончательной верификации диагноза требуется молекулярно-генетическое исследование, для выполнения которого требуется специализированное оборудование и обученный персонал, что влечет за собой большие финансовые затраты и пока не позволяет широко применять этот метод в клинической практике [3, 18]. Вместе с этим важным моментом является оценка степени выраженности фиброза печени у детей с ГБ. При рутинном ультразвуковом исследовании невозможно разграничить промежуточные стадии печеночного фиброза и не всегда удается выявить начальные признаки цирроза. Основным методом диагностики выраженности патологических изменений в печени является пункционная биопсия [27]. Однако ее применение в педиатрической практике ограничено ввиду инвазивности процедуры, необходимости анестезиологического пособия, проведение которого не всегда возможно детям с тяжелым течением ГБ из-за выраженной метаболической декомпенсации и высокого риска развития осложнений. В последние годы активно обсуждается роль сывороточных маркеров фиброза печени. Наиболее изученные – гиалуроновая кислота, коллаген IV типа, матриксные металлопротеиназы и их тканевые ингибиторы. Была показана их информативность в диагностике различных стадий фиброза печени у детей с хроническими формами ее патологии, в т.ч. и при ГБ [28, 29].
Лечение
На сегодняшний день наиболее актуальной проблемой для пациентов с печеночными формами ГБ и лечащих врачей является отсутствие разработанной патогенетической терапии, что неизбежно отражается на прогнозе заболевания и качестве жизни больных. Поэтому основным методом лечения продолжает оставаться специфическая диетотерапия, главной целью которой является нормализация углеводного обмена и профилактика вторичных метаболических нарушений путем максимально возможного длительного поддержания нормогликемии. В этом плане большое значение придается организации дробного питания с равномерным распределением легкорастворимых углеводов в течение суток, с этой целью количество приемов пищи увеличивается до 6–8 раз в день (включая ранний завтрак и поздний ужин). Неотъемлемой составляющей диеты является назначение сырого кукурузного крахмала, имеющего свойство медленно и непрерывно расщепляться под действием панкреатической амилазы до глюкозы, что продлевает период нормогликемии. Крахмал необходимо употреблять каждые 4–6 часов (включая ночной прием). Из пищевого рациона исключаются сахароза (пищевой сахар), фруктоза и галактоза, т.к. эти сахара у пациентов с ГБ обмениваются не до глюкозы, как у здоровых, а до лактата, что может усугублять лактат-ацидоз. Также строго ограничиваются содержащие сахар фрукты (свежие и сушеные), все сладкие кондитерские изделия, соки, фруктовые воды, мед, некоторые медикаменты. Разрешаются зеленые яблоки, лимон, варенье, приготовленное на глюкозе, из овощей – капуста, шпинат, лук-порей [30].
В качестве гепатопротекторной терапии могут быть использованы препараты урсодеоксихолевой кислоты (например, Урсофальк, Урсосан) и эссенциальные фосфолипиды (например, Эссенциале-форте Н). При вторичных осложнениях ГБ проводится коррекция тубулярных расстройств, остеопороза, устранение застойных явлений в желчном пузыре. Основным лечебным мероприятием при возникновении метаболического ацидоза является в/в введение щелочных растворов натрия гидрокарбоната. При нейтропении к детям с ГБ Ib-типа применяется гранулоцитарный колоние-стимулирующий фактор [26].
Перспективным направлением экспериментальной медицины за рубежом в настоящее время служит исследование эффективности аденовирусных векторов в качестве потенциальной патогенетической ферментозаместительной терапии при печеночных формах ГБ, которое, однако, пока проводится только на животных моделях [31].
За последние годы появились данные об успешной трансплантации печени небольшому числу пациентов с ГБ [32]. В нашей стране за 2009–2011 гг.
первые подобные операции были проведены трем детям с I типом этой патологии. Дальнейшее наблюдение за пациентами показало положительные результаты: нормализовались лабораторные показатели, случаев гипогликемии не фиксировалось, нивелировался лактат-ацидоз. Таким образом, первый успешный клинический опыт свидетельствует о возможности выполнения трансплантации фрагментов печени от живых родственных доноров и открывает перспективы для радикального лечения пациентов с гликогенозом I типа в России [33, 34].
В настоящее время благодаря развитию эффективных диагностических и лечебных подходов большинство хронических заболеваний можно реально контролировать и обеспечивать профилактику осложнений. Полноценное и своевременное применение этих мер существенно продлевает жизнь больных, а также повышает ее качество. Однако успешно контролировать хроническое заболевание без активного участия самого пациента не представляется возможным. Владение навыками контроля и лечения своего заболевания требует специальной подготовки с участием специалистов, которую называют обучением больных [35]. Как и при сахарном диабете, при ГБ чрезвычайно важна необходимость самоконтроля. С помощью современных методов экспресс-анализа (глюкометры, тест-полоски) больные самостоятельно могут оценивать важнейшие параметры обмена веществ с точностью, близкой к лабораторной. Целями самоконтроля служат профилактика осложнений, нормальное самочувствие, нормальное физическое развитие и рост, полноценная жизнь в обществе и высокое качество жизни. Следует помнить, что основной смысл самоконтроля – не только регулярная проверка показателей гликемии, но и адекватная оценка результатов, самостоятельная коррекция лечения и планирование определенных действий, если результаты самоконтроля неудовлетворительны [36, 37].
В настоящее время в нашей стране терапевтическое обучение пациентов с ГБ не разработано, несмотря на то что это является чрезвычайно актуальной проблемой отечественного здравоохранения. В связи с этим следует отметить, что любое хроническое заболевание снижает качество жизни ребенка, а также семьи, в которой он воспитывается [38]. Поэтому процесс реабилитации детей с ГБ должен подразумевать и оценку данного показателя. В последнее время это понятие стало неотъемлемой частью здравоохранения и прочно вошло в клинические и медико-социальные исследования. В зарубежной педиатрии показатель качества жизни включен в стандарты обследования больных, активно используется в популяционных исследованиях для разработки возрастно-половых нормативов, осуществления индивидуального мониторинга различных контингентов детей, оценки эффективности лечения, профилактических и реабилитационных мероприятий, прогноза заболевания, а также определения комплексного влияния хронической патологии на детей [39]. Качество жизни – это многогранное понятие, с помощью которого исследователи пытаются измерить оценку людьми собственного состояния. Оно охватывает физическое, психологическое и социальное благополучие так, как его воспринимает сам пациент и позволяет количественно оценить влияние на перечисленные составляющие болезней, травм и методик лечения [40]. В ряде научных исследований было показано, что качество жизни хронически больных людей по всем параметрам ниже, чем здоровых [41]. При этом существуют и особенности нарушения этого показателя в зависимости от нозологии как по степени снижения, так и по нарушению отдельных составляющих качества жизни [42]. Кроме того, было установлено, что качество жизни в период обострения заболевания значительно ухудшается по сравнению с ремиссией, а также зависит от частоты обострений [43]. В то же время необходимо отметить, что качество жизни не всегда идентично тяжести и длительности заболевания [44]. Так, в дебюте болезни все составляющие качества жизни могут сильно снижаться в результате стресса от факта установления диагноза, а затем больной привыкает к наличию хронического заболевания и его качество жизни может снова повыситься. Помимо этого качество жизни оценивает, как именно больной переносит свое состояние: оптимистично настроенные люди имеют более высокие показатели, чем пессимисты [45].
Нами было установлено, что у детей с ГБ отмечается резкое ограничение физической, психической и социальной адаптации, а также изменяется процесс межличностного взаимодействия больного ребенка с окружающими, поскольку он часто не может посещать детские коллективы, полноценно общаться со сверстниками. Постоянное эмоциональное напряжение приводит к социально-психологической дезадаптации, что значительно снижает качество жизни больных ГБ [46]. Традиционные методы обследования дают одностороннее представление о болезни и эффективности лечения, но не позволяют оценивать психологическую, социальную дезадаптацию ребенка, его отношение к своему состоянию [47]. Исследование качества жизни детей открывает возможность полноценного комплексного анализа состояния здоровья ребенка и повышает эффективность оказываемой ему медицинской помощи. Информация о параметрах качества жизни ребенка может представлять ценность не только для педиатров, психологов и других специалистов, но и для родителей ребенка. Включение оценки качества жизни в программу комплексной реабилитации детей с ГБ может решить эту проблему, что позволит повысить качество медицинской и социальной помощи этой группе больных [48].
Таким образом, подводя итог вышесказанному, актуальными проблемами для детей с ГБ являются повышение качества диагностики этой патологии врачами поликлиник, своевременное направление таких пациентов в специализированные медицинские учреждения для назначения комплексного лечения и последующего регулярного динамического наблюдения с целью оценки эффективности проводимой терапии и своевременной ее коррекции в случае необходимости. Чрезвычайно важна организация индивидуального и группового терапевтического обучения детей с ГБ в амбулаторных и стационарных условиях с изданием специализированной литературы для врачей, медицинских сестер, пациентов и их родителей. Необходимо беспрепятственное обеспечение пациентов с ГБ по месту жительства необходимыми лекарственными препаратами и индивидуальными средствами самоконтроля (глюкометры, тест-полоски). Требуются активные научные исследования, направленные на разработку эффективного патогенетического лечения ГБ в России.
Гликогеноз I типа (болезнь Гирке)
Клинические проявления гликогеноза типа I у новорожденных, грудных детей и детей старшего возраста неодинаковы. Причина — различия рациона и режима питания в этих возрастных группах.
Иногда в первые дни и недели жизни возникает гипогликемия голодания, однако в большей части случаев болезнь протекает бессимптомно, поскольку грудной ребенок часто питается и получает достаточное количество глюкозы. Нередко болезнь диагностируют через несколько месяцев после рождения, когда у ребенка обнаруживают увеличение живота и гепатомегалию. Бывают одышка и субфебрильная температура без признаков инфекции. Одышка вызвана гипогликемией и лактацидозом из-за недостаточной продукции глюкозы. Когда интервалы между кормлениями увеличиваются и ребенок начинает спать ночью, появляются симптомы гипогликемии, особенно по утрам. Тяжесть и длительность гипогликемии постепенно увеличиваются, что приводит к системным метаболическим нарушениям.
Если лечение не проводят, изменяется внешность ребенка. Характерны гипотрофия мышц и скелета, задержка роста и физического развития, отложение жира под кожей. Ребенок становится похож на больного с синдромом Кушинга. Развитие познавательных и социальных навыков не страдает, если только повторные приступы гипогликемии не вызвали повреждения головного мозга. Если ребенок не получает достаточного количества углеводов и гипогликемия голодания сохраняется, то задержка роста и физического развития становится резко выраженной. Некоторые дети с гликогенозом типа I умирают от легочной гипертензии.
Нарушение функции тромбоцитов проявляется повторными носовыми кровотечениями или кровоточивостью после стоматологических и других хирургических вмешательств. Отмечаются нарушения адгезии и агрегации тромбоцитов; нарушено также высвобождение АДФ из тромбоцитов в ответ на адреналин и контакт с коллагеном. Тромбоцитопатия вызвана системными метаболическими нарушениями; после лечения она исчезает.
УЗИ и экскреторная урография выявляют увеличение почек. У большинства больных выраженных нарушений функции почек не бывает, отмечается лишь повышение СКФ (скорость клубочковой фильтрации) . В очень тяжелых случаях может развиться тубулопатия с глюкозурией, фосфатурией, гипокалиемией и аминоацидурией (как при синдроме Фанкони). У подростков иногда наблюдается альбуминурия, а у молодых людей часто развивается тяжелое поражение почек с протеинурией, повышением АД (артериального давления) и падением клиренса креатинина, обусловленное фокально-сегментарным гломерулосклерозом и интерстициальным фиброзом. Эти нарушения приводят к терминальной почечной недостаточности.
Селезенка не увеличена.
Без лечения резко возрастают уровни свободных жирных кислот, триглицеридов и апопротеина C-III, который участвует в транспорте триглицеридови богатых триглицеридами липопротеидов. Уровни фосфолипидов и холестерина повышаются умеренно. Очень высокий уровень триглицеридов обусловлен их чрезмерной продукцией в печени и снижением их периферического метаболизма из-за снижения активности липопротеидлипазы. При тяжелой гиперлипопротеидемии на разгибательных поверхностях конечностей и ягодицах могут появляться эруптивные ксантомы.
Отсутствие лечения или неправильное лечение приводят к задержке роста и полового развития.
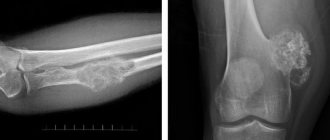
Аденомы печени по неизвестным причинам возникают у многих больных, обычно в возрасте 10-30 лет. Аденомы могут малигнизироваться, возможны кровоизлияния в аденому. На сцинтиграммах печени аденомы выглядят как участки пониженного накопления изотопа. Для обнаружения аденом применяют УЗИ. При подозрении на злокачественный рост более информативны МРТ (магнитно-резонансная томография) и КТ (компьютерная томография), позволяющие проследить превращение небольшого четко отграниченного новообразования в более крупное, с размытыми краями. Рекомендуется периодически измерять уровень альфа-фетопротеина в сыворотке (это маркер печеночноклеточного рака).
С возрастом тяжесть гипогликемии голодания уменьшается. Вес тела растет быстрее, чем вес головного мозга, поэтому соотношение между скоростью продукции и утилизации глюкозы становится более выгодным. Скорость продукции глюкозы возрастает за счет активности амило-1,6-глюкозидазы в печени и мышцах. В результате уровень глюкозы натощак постепенно повышается.
Клинические проявления гликогеноза типа Iа и типа Ib одинаковы, но при гликогенозе типа Ib наблюдается постоянная или преходящая нейтропения. В тяжелых случаях развивается агранулоцитоз. Нейтропения сопровождается дисфункцией нейтрофилов и моноцитов, поэтому повышается риск стафилококковых инфекций и кандидоза. У некоторых больных возникает воспалительное заболевание кишечника , напоминающее болезнь Крона.
Газета «Новости медицины и фармации» Гастроэнтерология (304) 2009 (тематический номер)
К патологии печени приводит гетерогенная группа наследственных болезней, обусловленных различными видами нарушения обмена углеводов. Различают:
— нарушения обмена моно- и ди-сахаридов; — болезни накопления — гликогенозы; — патология соединительной ткани — мукополисахаридозы; — другие.
Нарушения обмена моно- и дисахаридов
Фруктоземия
Этиология и патогенез.
Заболевание обусловлено врожденным отсутствием ферментов фруктозофосфатальдолазы и фруктозодифосфатальдолазы. Избыточное накопление фруктозофосфата нарушает гликогенолиз, что приводит к гипогликемии. В печени имеется недостаточное количество фермента фруктозо-1-фосфат-альдолазы, в результате продукты обмена (фруктозо-1-фосфат) накапливаются в организме (печени, почках, слизистых оболочках кишечника) и оказывают повреждающее действие. Морфологически в печени выявляются жировая инфильтрация, умеренный перилобулярный фиброз.
Клиническая картина.
Симптомы возникают при введении в рацион сладкой пищи или фруктовых соков, т.е. продуктов, содержащих фруктозу. Со 2–4-го месяца развиваются диспептические явления и состояния острой гипогликемии, которые проявляются бледностью, вялостью, потливостью, запахом ацетона. В тяжелых случаях может развиться гипогликемическая кома с потерей сознания и судорогами. Характерно, что гипогликемия возникает после приема пищи. С возрастом дети сами отказываются от сладкой пищи. Постоянным признаком является гепатомегалия (обычно с увеличением обеих долей) с ровным краем и некоторым уплотнением печени.
Лечение
. Диета, лишенная фруктозы, главными источниками которой считаются мед, сахарный тростник и свекла, фрукты, джемы, повидло, морковь, какао, цикорий, репа. Больным разрешается употреблять молоко и молочные продукты, яйца, маслины, подсолнечное масло, животные жиры. Разрешается женское и коровье молоко. Сухое молоко должно быть без сахара (сахарозы). Допускаются все виды сыров и натуральные кисломолочные продукты (неподслащенные). Запрещается молоко с добавлением сахарозы, сгущенное молоко, подслащенные кисломолочные продукты. Разрешены мясо и рыба. Исключаются колбасы и колбасные изделия, консервы. Жиры включаются в диету практически без ограничений (сливочное и растительное масло, маргарин). Почти все фрукты запрещены. Можно употреблять в пищу лимоны и каштаны. Из овощей допускаются зеленые бобы, кресс-салат, латук, лук-порей, капуста, шпинат. Разрешаются натуральная пшеничная или ржаная мука, рис, хлеб, манная крупа, чай, кофе, какао без сахара, глюкоза, мальтоза, декстрин-мальтоза, сахарин. Запрещаются соя, мука с сахарозой, бисквиты, пирожные, лимонад и все газированные фруктовые напитки, соки, сиропы, сахар, варенье, нуга, конфеты, все медикаменты, содержащие сахар, сорбитол (гранулы, драже, порошки, пилюли).
Новорожденным назначают молочные смеси без сахара. Дети первого года получают молочные смеси, содержащие только лактозу и декстрин-мальтозу. Вместо фруктовых пюре и соков питание дополняют глюкозой (от 30 до 60 г). При введении прикорма раньше, чем здоровым детям, назначают мясо, рыбу, сыр, яйца. Аскорбиновую кислоту используют без сахара. Диетическое питание показано до 5–6 лет, только по достижении этого возраста можно в ограниченных количествах, под контролем врача и биохимических анализов крови, включать в питание ребенка продукты из запрещенного списка.
Галактоземия
Частота 1 : 35 000–50 000 населения.
Этиология и патогенез
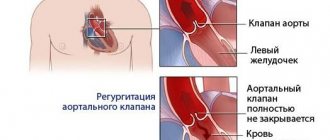
. Наследственная энзимопатия. Наследуется по рецессивному типу. В основе галактоземии (рис. 1) лежит нарушение обмена галактозы в связи с отсутствием фермента галактозофосфат-уридилтрансферазы. В результате в крови накапливаются в больших концентрациях галактоза и галактозофосфат. Происходит нарушение процесса ферментативного превращения галактозы в глюкозу с накоплением галактозы и продуктов ее обмена в клетках, что оказывает повреждающее действие на функции печени, головного мозга, хрусталика глаза, почек.
Клиническая картина
. Клинические признаки заболевания возникают рано — через 1–2 недели после рождения ребенка. Пропадает аппетит, появляются вялость, рвота, понос. Наблюдается дефицит массы тела. Постепенно развивается гепато-, спленомегалия, появляется стойкая гипербилирубинемия, преимущест-венно за счет прямого билирубина. Часто отмечается катаракта, ведущая к слепоте. Могут быть симптомы, свидетельствующие о поражении почек (протеинурия, гипераминоацидурия), центральной нервной системы (задержка психофизического развития). После чайно-водной паузы состояние улучшается, но введение молока обусловливает рецидив нарушений со стороны желудочно-кишечного тракта. При несвоевременной диагностике заболевание прогрессирует, что приводит к тяжелым последствиям или летальному исходу.
Диагностика:
1. Определение концентрации га-лактозо-1-фосфата в эритроцитах (повышена). 2. Исследование активности галак-тозо-1-фосфат-уридилтрансферазы в эритроцитах. 3. Повышение уровня галактозы в крови и моче (методом хроматографии). 4. Микробиологический тест Гатри.
Лечение.
Диетотерапия является единственным методом лечения. Для вскармливания ребенка используют смеси, лишенные лактозы. Из питания детей более старшего возраста исключают цельномолочные продукты.
Гликогенозы
Этиология и патогенез
. Группа наследственных болезней обмена полисахаридов, развивающихся в результате нарушения синтеза или распада гликогена на простые сахара. При этом нормальный и аномальный гликоген одновременно накапливаются в клетках печени и других органах и тканях.
Гликоген — это сильно разветвленный полимер глюкозы, в котором большинство остатков имеют 1,4-связи, а 7–10 % остатков — 1,6-связи. Древовидная структура подвергается надстройке и отщеплению остатков на периферии молекулы. Молекулярная масса гликогена составляет несколько миллионов, его молекулы могут агрегировать с образованием структур, видимых при электронной микроскопии. В печени гликогена обычно содержится менее 70 мг/г, а в мышцах — менее 15 мг/г, но эти величины колеблются в зависимости от питания и гормональных влияний. Нарушения структуры гликогена могут быть связаны как с уменьшением, так и с увеличением ветвления молекулы.
Метаболические пути синтеза и распада гликогена в разных тканях различны; например, некоторые реакции активно протекают в печени, но менее активны или отсутствуют в мышцах, а ряд ферментов в мышцах и печени кодируются разными генами. Глюкоза плазмы проникает в клетку и фосфорилируется глюкокиназой или гексокиназой. Первая содержится в печени, осуществляющей фосфорилирование основной массы глюкозы, тогда как многочисленные гексокиназы распределены по тканям более широко. Глюкозо-6-фосфат (Г-6-Ф) превращается в глюкозо-1-фосфат (Г-1-Ф) в обратимой реакции, катализируемой фосфоглюкомутазой. Уридиндифосфатглюкоза (УДФГ) синтезируется из Г-1-Ф и уридинтрифосфата под действием УДФГ-пирофосфорилазы. Генетическая недостаточность ни одного из этих печеночных ферментов не была зарегистрирована. Молекула гликогена затем удлиняется путем присоединения отдельных остатков глюкозы из УДФГ, в результате чего образуется полимер. Эта реакция катализируется гликогенсинтазой, которая существует в активной дефосфорилированной и неактивной фосфорилированной формах. Для синтеза нормально разветвленной молекулы гликогена требуется также участие ветвящего (бранчер) фермента (1,4-a-гликан: 1,4-a-гликан-6-гликозилтрансфераза), который пе-реводит 1,4-связи олигосахарида в положение 1,6-связей.
Глюкоза мобилизуется из гликогена целым рядом ферментативных реакций. На гликоген непосредственно действует активная форма фосфорилазы — фосфорилаза а, отщепляя отдельные остатки глюкозы и образуя Г-1-Ф. В печени и мышцах фосфорилаза кодируется разными генными продуктами. В этих тканях фермент может существовать в активной фосфорилированной и неактивной дефосфорилированной формах. Фосфорилаза представляет собой димер, состоящий из одинаковых субъединиц, причем обе формы фермента подвергаются сложной аллостерической регуляции. Неактивная фосфорилаза b превращается в активную форму под действием фосфорилазо-b-киназы, которая существует и в активной фосфорилированной и неактивной дефосфорилированной формах. Этот фермент состоит из четырех разных субъединиц (a, b, g, d4),причем d-цепь идентична связывающему кальций белку — кальмодулину. Скорость мобилизации глюкозы этой системой регулируется каскадом киназных реакций, включающих цАМФ.
По характеру ферментной недостаточности принято различать 12 типов гликогенозов, среди которых выделяют печеночные (гликогенозы 0, I, III, IV, VI, VIII, IX, Х, ХI типов) или мышечные формы (гликогенозы V и VII типов). Гликогеноз II типа проявляется поражением только мышц или поражением многих систем и органов (генерализованная форма). Также возможны сочетания нескольких типов.
Клиническая картина
гликогенозов характеризуется гипогликемией (рвота, судороги, потеря сознания, кома). Течение болезни зависит от места депонирования гликогена: печень, почки, мышечная ткань. Соответственно выделяют цирроз печени, почечную форму, мышечный синдром (включая сердечную форму). Преобладание у новорожденного ребенка симптомов гипогликемии может привести к синдрому внезапной смерти. Прогноз зависит от типа болезни.
Классификация основана на различиях в дефектах ферментов, лежащих в основе заболеваний.
Гликогеноз 0 типа
(агликеноз) характеризуется резким снижением запасов гликогена в печени, наблюдается тяжелое состояние вплоть до развития комы (гипогликемический синдром). Кома может возникать после рождения при позднем прикладывании ребенка к материнской груди, а позднее — утром натощак и в перерывах между кормлениями. При отсутствии лечения ребенка наступает нарушение психомоторного развития.
Гликогеноз I типа
(болезнь Гирке) (рис. 2) — гликогеноз, обусловленный недостаточностью глюкозо-
6-фосфатазы, приводящей к невозможности превращения глюкозо-6-фосфата в глюкозу, что сопровождается накоплением гликогена в печени и почках; наследуется по аутосомно-рецессивному типу. Дефект фермента в печени, почках, слизистой оболочке тонкой кишки. При его первых проявлениях наблюдаются отсутствие аппетита, рвота, респираторный дистресс-синдром, гипогликемические судороги (кома), которые выявляются сразу после рождения или в грудном возрасте. Прогрессируют гепатомегалия и нефромегалия за счет гликогенной инфильтрации. С течением времени появляются: отставание в росте, диспропорция тела (большая голова, короткие шея и ноги), кукольное лицо, гипотония мышц; половое созревание задерживается. Нервно-психическое развитие удовлетворительное. В связи с резкой гипогликемией больные вынуждены постоянно принимать пищу.
Гликогеноз II типа
(болезнь Помпе) — наследуется по аутосомно-рецессивному типу. Симптомы проявляются в первые недели жизни — до шести месяцев после рождения. Дефект фермента найден в печени, почках, селезенке, мышцах, нервной ткани, лейкоцитах. Наблюдается расстройство дыхания, беспокойство или адинамия. Отмечаются отсутствие аппетита, задержка роста, мышечная гипотония. Увеличиваются размеры сердца, печени, почек, селезенки. Сердце приобретает шаровидную форму, в связи с гипертрофией миокарда появляются изменения ЭКГ. Часто возникают гипостатические пневмонии, бронхиты, ателектазы легких, наблюдаются миодистрофия, гипорефлексия, спастические параличи. Мышечная форма гликогеноза II типа возникает только в мышцах при дефиците кислой α-1,4-глюкозидазы. Болезнь проявляется в более поздние сроки и по клинической картине напоминает миопатию.
Гликогеноз III типа
(болезнь Кори, болезнь Форбса, лимитдекстриноз) — гликогеноз, вызванный недостаточностью фермента амило-1,6-глюкозидазы. Этот фермент катализирует расщепление связей […C-O-C…] в молекуле гликогена в точках ветвления. Болезнь сопровождается отложением атипичного гликогена в печени, сердце, мышцах. Наследуется по аутосомно-рецессивному типу. Дефект фермента найден в печени, мышцах, лейкоцитах, эритроцитах. С первых месяцев жизни ребенка наблюдаются гепатомегалия, мышечная гипотония, гипертрофия отдельных групп мышц. В некоторых случаях у больных отмечаются нарушение сердечной проводимости и кровообращения, гипертрофия миокарда. Развитие заболевания замедляется после пятилетнего возраста или в пубертатном периоде.
Гликогеноз IV типа
(болезнь Андерсена, амилопектиноз, диффузный гликогеноз с циррозом печени) — гликогеноз, семейный цирроз печени, вызванный дефектом фермента амило-(1,4-1,6)-трансглюкозидазы. Этот фермент катализирует превращение 1,4-связей в молекуле гликогена в 1,6-связи, то есть обусловливает ветвление молекулы полисахарида. Заболевание сопровождается избыточным накоплением атипичного гликогена в печени. Наследуется по аутосомно-рецессивному или связанному с полом типу. Дефект фермента найден в печени, почках, мышцах, лейкоцитах. Болезнь наблюдается с первых месяцев жизни и характеризуется гепатоспленомегалией, развитием цирроза печени, желтухой, гипогликемией.
Гликогеноз V типа
(болезнь Мак-Ардла, миофосфорилазная недостаточность) — гликогеноз, связанный с дефектом мышечной фосфорилазы. Заболевание, обусловленное нарушением каталитической функции этого фермента; сопровождается отложением значительного количества гликогена в мышцах. Наследуется по аутосомно-рецессивному типу. Дефект фермента найден в мышцах. В связи с гликогенной инфильтрацией скелетные мышцы увеличиваются в объеме, становятся очень плотными. Мышечная слабость, мышечные спазмы, тахикардия при физической нагрузке появляются в первые десять лет жизни и прогрессируют. Наблюдается транзиторная миоглобинурия. Концентрация лактата в крови после физической нагрузки уменьшается. Чаще (в 5 раз) болеют лица мужского пола.
Гликогеноз VI типа
(болезнь Герса, гепатофосфорилазная недостаточность) — гликогеноз, вызванный недостаточностью фосфорилазы печени. Фосфорилаза печени катализирует фосфорилирование гликогена с образованием глюкозо-1-фосфата. Нарушение этого механизма приводит к избыточному отложению гликогена в печени. Наследуется, предположительно, по аутосомно-рецессивному типу. Проявляется обычно на первом году жизни. Характерны значительное увеличение печени в результате гликогенной инфильтрации гепатоцитов, задержка роста, кукольное лицо, гиперлипидемия, гипергликемия после внутривенного введения галактозы, повышенное содержание гликогена в эритроцитах. Наследуется по аутосомно-рецессивному типу. Дефект фермента найден в печени, лейкоцитах. Проявляется обычно на первом году жизни.
Гликогеноз VII типа
(болезнь Таруи, миофосфофруктокиназная недостаточность) — симптомы сходны с гликогенозом V типа. Дефект фермента найден в мышцах, эритроцитах. Также характерны мышечная слабость, утомляемость и отсутствие гиперлактацидемии после физической нагрузки.
Гликогеноз VIII типа
(болезнь Томсона) — встречается редко, наследование не установлено. Дефект фермента найден в печени, в головном мозге. После рождения увеличиваются размеры печени, появляются нистагм («танцующие глаза») и атаксия. Неврологическая симптоматика прогрессирует.
Гликогеноз IX типа
(болезнь Хага) — наследуется по рецессивному, связанному с полом типу. Дефект фермента найден в печени. У больных наблюдается гепатомегалия.
Гликогеноз Х типа
— известен случай у единственного больного, наследование не установлено. Дефект фермента найден в печени, мышцах. Наблюдалась гепатомегалия, через 6 лет после начала заболевания появились мышечные боли и спазмы мышц после физических упражнений.
Гликогеноз XI типа
(болезнь Фанкони — Бикеля) — наследование не установлено. Дефект фермента найден в печени, почках. Характеризуется значительным увеличением печени и резкой задержкой роста. Наблюдаются симптомы гипофосфатемического рахита. В пубертатном периоде возможны уменьшение размеров печени, ускорение роста, нормализация уровня фосфора в крови.
Лечение
гликогенозов в основном симптоматическое и направлено на изменение нарушений обменных процессов. Цель лечения — предупредить тяжелую гипогликемию. Назначают диету, богатую белками и углеводами. Питание должно быть частым (каждые 4 ч). Белки служат источником аминокислот — субстратов глюконеогенеза; они уменьшают углеводную нагрузку, приводящую к гипергликемии и лактат-ацидозу. Такая диета предотвращает гипогликемию и кетоацидоз натощак, уменьшает гипергликемию и лактат-ацидоз после еды и способствует ускорению роста. При мышечных формах гликогенозов улучшение отмечается при соблюдении диеты с высоким содержанием белка, назначении фруктозы, поливитаминов, АТФ. Иногда необходимо применение глюкагона, анаболитических гормонов, глюкокортикоидов. Предпринимаются попытки введения больным недостающих ферментов.
Профилактика
не разработана.
Мукополисахаридозы
Этиология и патогенез
. Мукополисахаридозы (МПС) — гетерогенная группа заболеваний, отнесенных к наследственным болезням обмена сложных сахаров. МПС сопровождаются избыточным накоплением в тканях и повышенной экскрецией гликоз-аминогликанов (ГАГ) — кислых мукополисахаридов, соединенных с белком и состоящих из уроновых кислот, аминосахароз и нейтральных сахаров. Указанные комплексы существуют в форме протеогликанов, являющихся важнейшими компонентами основного структурного белка волос (0-кератина) и структурного белка соединительной ткани (коллагена).
Для большинства МПС характерен аутосомно-рецессивный тип наследования, кроме синдрома Хантера (Х-сцепленный рецессивный).
Клиническая картина
. Манифестация болезни, как правило, в возрасте до 7 лет, задержка роста (до карликовости), контрактура суставов, кифоз/кифосколиоз/сколиоз, массивный череп с глубоким и удлиненным турецким седлом, короткая шея, деформация грудной клетки, веслообразные ребра, укорочение трубчатых костей, грубые черты лица, помутнение роговицы, гепатоспленомегалия, задержка психического развития, судороги, глухота, грыжи (пупочная, паховая, пахово-мошоночная), врожденные пороки сердца, гликозаминогликанурия (100–200 мг в сутки). Характерные для МПС признаки дисморфизма получили название «гаргоилический фенотип» (гаргоилизм — синоним МПС).
Среди МПС выделяют ряд типов, каждый из которых обусловлен дефицитом специфической лизосомной гидролазы, участвующей в последовательном расщеплении ГАГ.
I тип — синдром Гурлера (4р16 — IDUA). Выделяют подтипы: Гурлер, Шейе, смешанный вариант. Для всех характерно снижение активности альфа-идуронидазы и накопление в тканях дерматан- и гепарансульфатов.
II тип — синдром Хантера (Гюнтера) (Хq28 — IDS). Снижение активности L-идуросульфат-сульфатазы и отложение в тканях дерматан- и гепарансульфатов. Клинические признаки менее выражены, продолжительность жизни больше, чем при других типах МПС.
III тип — синдром Санфилиппо (12q13.4). В зависимости от природы первичного биохимического дефекта выделяют четыре подтипа: А, В, С, D. В клинической картине преобладают психические расстройства: деменция, агрессивность. Продолжительность жизни не превышает 20 лет.
IV тип — синдром Моркио (16q24.3 — GALNS). Выделяют подтипы А и В. В тканях откладывается кератансульфат. Преобладают поражения скелета и непропорционально низкий рост.
V тип — синдром Шейе (см. I тип).
VI тип — синдром Марото — Лами (5q11.2 — ARSB). Дефицит фермента арилсульфатазы В. В тканях накапливается дерматансульфат. Фенотипически напоминает МПС I типа, но интеллект не снижен.
VII тип — синдром Слая (7q21.11 — GUSB). Дефицит фермента δ-глюкуронидазы. В тканях накапливаются дерматан-, гепаран- и хондроитин-сульфаты. Фенотипиче-ски напоминает МПС I типа, но имеет более доброкачественное течение.
Диагностика МПС
основывается на совокупности данных генеалогического анализа, клинических проявлений, типичных рентгенологических данных, экскреции с мочой оксипролина (снижение), ГАГ и их фракций (превышение в 5–10 раз). Точная идентификация типов МПС возможна только с помощью определения активности лизосомных гидролаз в лимфоцитах и лейкоцитах крови, культуре фибробластов кожи, биоптатов печени, а также в моче.
Лечение
больных МПС в основном симптоматическое, заключается в назначении терапии, способствующей нормализации или стабилизации патологического процесса в опорно-двигательном аппарате, сердечно-сосудистой и центральной нервной системах, паренхиматозных органах, органах зрения и слуха. Перспективным считается плазмаферез.
Публикации в СМИ
Гликогенозы — группа наследственных заболеваний, вызванных недостаточностью одного или нескольких ферментов, вовлечённых в синтез и распад гликогена, и характеризующихся накоплением патологических количеств или типов гликогена в тканях. Симптоматика возникает вследствие накопления гликогена или его промежуточных метаболитов или из-за недостатка конечных продуктов распада гликогена, особенно глюкозы. Гликоген и некоторые из промежуточных метаболитов, депонированных в тканях, могут быть обнаружены при МРТ. Различия в степени тяжести и возрасте начала клинических проявлений вызваны вовлечением различных изоферментов или других компонентов повреждённых ферментных систем. Частота всех форм болезней накопления гликогена — 1:40 000 населения.
Генетическая классификация и клиническая картина
• Гликогеноз типа 0 (*240600, 12p12.2, ген GYS2 [138571], r) — недостаточность гликоген синтетазы (КФ 2.4.1.11) печени. Клиническая картина: гипогликемия и гиперкетонемия натощак, судороги, гипергликемия и гиперлактатемия после приёма пищи.
• Гликогеноз типа I(a) (232200, 17q21, Â) — недостаточность глюкозо-6-фосфатазы (КФ 3.1.3.9), приводящая к избыточному накоплению гликогена нормальной химической структуры (особенно в печени и почках). Наблюдают значительно чаще других гликогенозов. Клиническая картина: гипогликемия, артериальная гипертензия, задержка роста, позднее половое созревание, увеличение живота, аденомы печени, печёночноклеточная карцинома, гепатобластома, увеличение печени, хронический панкреатит, ксантомы, паукообразные гемангиомы, подагрические тофусы, протеинурия, гематурия, почечная недостаточность, центральный сегментарный гломерулосклероз, мочекислые камни почек, подагрический артрит, геморрагический диатез, лёгочная артериальная гипертензия. Лабораторные данные: недостаточность глюкозо-6-фосфатазы, гиперлипидемия, гиперурикемия, гиперлактацидемия, кетонемия, метаболический ацидоз. Синонимы: фон Гирке болезнь, нефромегалический гликогеноз, фон Гирке ван Кревельда синдром, фон Гирке ван Кревельда болезнь.
• Гликогеноз типа Ib (232220, r) — мутации гена транспортёра глюкозо 6 фосфата (*602671, 11q23.3). Клиническая картина: диарея, плохой аппетит, болезнь Крона, хронические остеомиелиты, перианальные абсцессы, нейтропения, гипохромная анемия, тромбоцитопения, вторичный амилоидоз, протеинурия, гиперлипидемия.
• Гликогеноз типа Ic (232240, r) — дефект транспортёра глюкозо 6 фосфата (*602671, 11q23.3). Клиническая картина: гипогликемия, артериальная гипертензия, протеинурия, гематурия, почечная недостаточность, центральный гломерулосклероз, задержка роста и полового созревания, опухоли печени (аденомы, печёночноклеточная карцинома, гепатобластома), увеличение печени, хронический панкреатит, ксантома, ангиома кожи, подагрические тофусы, подагрический артрит, нормальные функции лейкоцитов, лёгочная артериальная гипертензия. Лабораторные данные: дефицит фосфат пирофосфат транслоказы, гиперлипидемия, гиперурикемия, гиперлактацидемия, кетонемия, метаболический ацидоз. ЛС (аллопуринол) следует назначать с осторожностью. Течение обычно хроническое прогрессирующее.
• Гликогеноз типа IIa (153360 — недостаточность лизосомной a-1,4 глюкозидазы, приводящий к избыточному накоплению гликогена нормальной химической структуры в сердце, скелетных мышцах, печени, мозге. Клиническая картина: кардиомиопатия, кардиомегалия, артериальная гипотензия, миотония, мышечная слабость, утомляемость, увеличенный язык, смерть на первом году жизни, дыхательная недостаточность, одышка, аневризмы мозговых артерий. Синоним: Помпе болезнь.
• Гликогеноз типа IIb (300257, дефект ассоциированного с лизосомами мембранного белка LAMP2, r). Клиническая картина: слабость проксимальных мышц, гипертрофическая кардиомиопатия, сердечная недостаточность, АВ-блокада, умственная отсталость. Лабораторные данные: накопление гликогена в лизосомах; активность кислой мальтазы нормальная. Синонимы: болезнь Антополя, болезнь Данона (300257, дефект лизосомного белка LAMP2).
• Гликогеноз типа III (*232400, 1p21, ген AGL, GDE, r) — недостаточность амило-1,6-глюкозидазы, приводящая к накоплению гликогена ненормальной структуры с короткими внешними цепями в печени и мышцах. Клиническая картина: миопатия, увеличение печени, гипогликемия, кетоацидоз, мышечная слабость с атрофией мышц, «ангельское» лицо, склонность к кровотечениям (в т.ч. носовым), гипертрофия желудочков на ЭКГ. Синонимы: Кори болезнь, Форбса болезнь, лимитдекстриноз.
• Гликогеноз типа IV (*232500, 3p12, ген GBE1, r) — недостаточность 1,4-a-глюкан ветвящего фермента (КФ 2.4.1.18), приводящая к накоплению гликогена ненормальной структуры с длинными цепями в печени, почках, мышцах и других тканях. Клиническая картина: цирроз печени, портальная гипертензия, печёночная недостаточность, гипогликемия, кардиомиопатия, сердечная недостаточность, миопатия, тазово-плечевая мышечная дистрофия, гиперлордоз позвоночника, походка вразвалку, слабость проксимальных мышц конечностей, смерть до 4-летнего возраста. Синонимы: болезнь Андерсен, амилопектиноз.
• Гликогеноз типа V (*232600, 11q13, ген PYGM, r) — дефект амилофосфорилазы (КФ 2.4.1.1), вызывающий накопление гликогена нормальной химической структуры в мышцах. Клиническая картина: слабость и атрофия скелетных мышц, мышечные боли при нагрузке, миоглобинурия. Синонимы: МакАрдла–Шмида–Пирсона болезнь, миофосфорилазная недостаточность, МакАрдла болезнь.
• Гликогеноз типа VI (*232700, 14q21–q22, ген PYGL, r) — недостаточность амилофосфорилазы (КФ 2.4.1.1), приводящая к накоплению гликогена нормальной структуры в гепатоцитах и лейкоцитах. Клиническая картина: увеличение печени, кетоз, гипогликемия, задержка роста. Синонимы: Гирса болезнь, гепатофосфорилазная недостаточность.
• Гликогеноз типа VII (*232800, 12q13.3, ген PFKM, r) — миопатии и увеличение печени, обусловленные недостаточностью 6 фосфофруктокиназы (КФ 2.7.1.11). Клиническая картина: миопатия, увеличение печени, слабость мышц, крампи, гемолиз, лёгкая полицитемия, ретикулоцитоз, умеренная желтуха, желчнокаменная болезнь. Лабораторные данные: недостаточность фосфофруктокиназы мышц, миоглобинурия, гиперурикемия. Синонимы: гепатофосфоглюкомутазная недостаточность, Томсона болезнь.
• Гликогеноз типа VIII (*261750, Xp22.2 p22.1, ген PHKA2, PHK; *311870, Xq12 q13, ген PHKA1, À) — недостаточность киназы фосфорилазы (КФ 2.7.1.38) в мышцах. Клинические и биохимические расстройства исчезают с возрастом, у большинства взрослых пациентов заболевание протекает бессимптомно. Клиническая картина: увеличение печени, задержка роста, почечный канальцевый ацидоз. Лабораторные данные: недостаточность печёночной киназы фосфорилазы (PHK); мышечная киназа фосфорилазы в пределах нормы; повышенное содержание глутамат-пируват и глутамат оксало-ацетат трансаминаз; гиперхолестеринемия; гипертриглицеридемия; кетонемия на фоне голодания; гиперлактацидемия или гиперурикемия отсутствует. Синонимы: Таруи болезнь, недостаточность киназы фосфорилазы печени и мышц. Примечание: некоторые субъединицы фермента имеют локусы в 16p12.1 p11.2.
• Гликогеноз типа VIIIb (261740, r) — крайне редкая форма недостаточности фосфофруктокиназы (КФ 2.7.1.38), ограниченная мышцей сердца.
• Гликогеноз типа VIIIc (*261750, r) — недостаточность киназы фосфорилазы (КФ 2.7.1.38) в печени и мышцах. Клиническая картина: увеличение печени, диарея, задержка роста, гипотония мышц, умеренная слабость. Лабораторные данные: недостаточность киназы фосфорилазы в печени и мышцах с накоплением гликогена.
ЛЕЧЕНИЕ • При типах 0, I и III — предотвращение гипогликемии и молочного ацидоза назначением дробных доз углеводов, что позволяет поддержать нормальные уровни глюкозы крови, предупредить развитие молочного ацидоза, гиперурикемии и гиперлипидемии. Кроме того, используют непрерывную подачу высокомолекулярных декстранов через эндоназальный зонд. Аллопуринол назначают для профилактики подагры и уратных камней почек • Ограничение анаэробной нагрузки уменьшает мышечные симптомы типов V и VII • При типе VIII рекомендуют ограничение физической нагрузки и обильное питьё • Эффективных методов лечения других типов нет.
МКБ-10 • E74.0 Болезни накопления гликогена
Болезнь Грейвса
Тиреотоксикоз
10385 10 Сентября
ВАЖНО!
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Болезнь Грейвса: причины появления, симптомы, диагностика и способы лечения.
Определение
Болезнь Грейвса (другие названия – Базедова болезнь, токсический зоб) – наследственное аутоиммунное заболевание, которое может развиться в любом возрасте. Женщины болеют примерно в 5 раз чаще, чем мужчины. Болезнь характеризуется избыточной выработкой тиреоидных гормонов, что приводит к тиреотоксикозу (избытку гормонов щитовидной железы в кровеносном русле).
Замечено, что патологический процесс часто запускается в ситуациях, когда организм испытывает стресс.
Причины появления болезни Грейвса
Болезнь Грейвса связывают с врожденным дефектом специфических Т-лимфоцитов (в организме Т-лимфоциты регулируют скорость защитной реакции, ее продолжительность, обеспечивают клеточный иммунитет). Вследствие срыва иммунологической толерантности происходит активация В-лимфоцитов, продуцирующих тиреостимулирующие иммуноглобулины – АТ-рТТГ. АТ-рТТГ воздействуют на рецепторы к ТТГ и начинают стимулировать клетки щитовидной железы. Это приводит к выраженному повышению выработки тиреоидных гормонов (Т4 и Т3), а также к увеличению щитовидной железы в размерах (гипертрофии).
Спусковым механизмом нарушения иммунологической толерантности может стать вирусная инфекция, стресс, курение.
При болезни Грейвса щитовидная железа может увеличиться до такой степени, что будет видна визуально. При отсутствии лечения она начинает сдавливать пищевод и трахею, вызывая нарушения глотания и дыхания.
Классификация заболевания
Классифицируется болезнь Грейвса по размеру зоба, а также по степени тяжести синдрома тиреотоксикоза.
В России до настоящего времени обычно используется классификация эндемического зоба по О.В. Николаеву, предложенная еще в 1955 году.
0-я степень – щитовидная железа не видна и не пальпируется;
1-я степень – щитовидная железа не видна, но пальпируется и виден при глотании перешеек;
2-я степень – щитовидная железа видна во время глотания, пальпируется, форма шеи не изменена;
3- я степень – щитовидная железа видна, меняется контур шеи («толстая шея»);
4-я степень – большой зоб, нарушающий конфигурацию шеи;
5-я степень – огромный зоб, сдавливающий пищевод и трахею.
Классификация по тяжести тиреотоксикоза
Субклинический период – заболевание устанавливается на основании данных гормонального исследования, клиническая картина может отсутствовать или быть стертой.
Манифестный период – присутствует развернутая клиническая картина заболевания.
Осложненный период – присутствуют осложнения (мерцательная аритмия, сердечная недостаточность, тирогенная относительная надпочечниковая недостаточность, дистрофические изменения паренхиматозных органов (печени, селезенки и т.д.), психоз, значительный дефицит массы тела).
Симптомы болезни Грейвса
Болезнь Грейвса отличается большим разнообразием клинических проявлений. Больные предъявляют жалобы на учащенное сердцебиение (тахикардию), аритмию, жар (непереносимость теплых и душных помещений), сильное потоотделение, ощущение внутренней дрожи, потерю массы тела при хорошем аппетите, частый неоформленный стул. Характерен подъем систолического («верхнего») давления и снижение диастолического (нижнего). Многие страдают из-за нарушения сна, быстрой утомляемости и снижения работоспособности. Отмечаются отеки ног, слабость мышц, неприятное чувство сжатости в области шеи.
Второе название заболевания – Базедова болезнь – свидетельствует о том, что у пациентов нередко регистрируются офтальмологические нарушения (эндокринная офтальмопатия), которые проявляются пучеглазием (поражением мягких тканей глазницы). Можно услышать жалобы на регулярное слезотечение, давление и «песок» в глазах, двоение, отечность век, светобоязнь.
При пальпаторном обследовании примерно у 80% пациентов удается выявить увеличенную щитовидную железу.
У пациентов репродуктивного возраста может наблюдаться снижение либидо (у мужчин), для женщин характерны нарушения менструального цикла.
Как правило, симптомы развиваются быстро, поэтому пациенты приходят на прием к врачу в течение 6-12 мес. после их манифестации.
Диагностика болезни Грейвса
При подозрении на наличие у пациента тиреотоксикоза следует исследовать уровень ТТГ высокочувствительным методом.