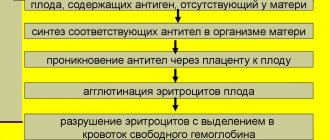
Талассемия – это генетическая болезнь крови, при которой, из-за мутации генов, образовывается недостаточное количество гемоглобина в организме и происходит деформация эритроцитов.
Гемоглобин – это специфический белок, который содержится в эритроцитах и отвечает за перенос железа и кислорода ко всем органам и тканям. Основная функция гемоглобина – дыхательная и его недостаток приводит к кислородному голоданию. В результате чего органы не могут полноценно выполнять свои физиологические функции.
Различают «взрослый» гемоглобин (HbA) и плодный тип гемоглобина (HbF).
Фетальный (плодный) гемоглобин синтезируется у плода спустя 2 недели после формирования внутренних органов (приблизительно с 12-й недели беременности). После рождения ребенка, HbF составляет до 80% и постепенно замещается «взрослым» HbA.
В норме у человека, плодного типа гемоглобина должно остаться не более 1,5%. Если случился сдвиг цифр влево или вправо – это всегда говорит о патологии (может быть нарушена работа сердечно-сосудистой системы, щитовидной железы, гипофиза, наличие гематологических отклонений, лейкоза, лимфогранулематоза, интоксикации).
Здоровая зрелая молекула гемоглобина (HbA) состоит из двух пар цепей – альфы и беты. Фетальный гемоглобин (HbF) состоит из цепей – гаммы и дельты.
А талассемия – это результат нарушенного синтеза одной или нескольких цепей гемоглобина. На основе чего различают альфа-талассемию, бета-талассемию, бета-дельту талассемию и т.д.
Тяжесть протекания того или другого вида заболевания зависит от количества поврежденных участков:
- Если изменен один ген цепи – заболевание (малая талассемия), протекает легко и бессимптомно. Но, при этом человек остается носителем данной болезни, которую может передать по наследству своим детям.
- Нарушение синтеза 2-х генов проявляется микроцитарной анемией в легкой степени тяжести.
- Патология 3-х генов протекает тяжело с ярко выраженной симптоматикой.
- Повреждение сразу 4-х генов (большая талассемия) – это наиболее редкий вид талассемии, который практически несовместим с жизнью (иногда ситуация решается пересадкой костного мозга).
Большая альфа-талассемия – самое серьезное отклонение, которое возникает еще в период внутриутробного развития плода. Грозит осложнениями, как для ребенка, так и для женщины.
Сегодня при своевременной внутриутробной трансфузии эритроцитарной массы удается сохранить жизнь плода.
Большая бета-талассемия (анемия Кули) – это также опасное нарушение, которое проявляется в первые два года жизни ребенка.
Малая альфа и бета-талассемия нарушает процесс эритропоэза (развитие новых эритроцитов), что приводит к хронической анемии и нарушению гемолиза.
Вне зависимости от вида, клиническое протекание талассемий схожи, различия зависят от степени тяжести патологического процесса.
Что такое талассемия и почему она возникает
Талассемия – это генетическое заболевание крови. Образуется недостаточное количество гемоглобина, белка, отвечающего за перенос кислорода и углекислого газа. Он состоит из четырех пептидов. 90% молекул гемоглобина содержат два альфа-глобина и два бета-глобина (гемоглобин А или A1), 2.5% содержат вместо бета-цепей дельта-цепи (гемоглобин А2), а остальные представляют собой гемоглобин A, состарившийся в процессе эксплуатации в эритроцитах (гемоглобин A3). При мутации в генах, отвечающих за синтез одного из глобинов, состав гемоглобина нарушается. Эти изменения влекут гибель эритроцитов.
Рекомендации
- ^ абc
«Бета-талассемия».
Домашний справочник по генетике
. Получено 2015-05-26. - ^ абc
Адвани, Пуджа. «Лечение и лечение бета-талассемии».
Medscape
. Получено 4 апреля 2021. - ^ аб
Маккинни, Эмили Слоун; Джеймс, Сьюзан Р .; Мюррей, Шэрон Смит; Нельсон, Кристина; Эшвилл, Джин (2014-04-17).
Уход за матерью и ребенком
. Elsevier Health Sciences. ISBN 9780323293778 . - Галанелло, Ренцо; Орига, Рафаэлла (21 мая 2010 г.). «Бета-талассемия». Орфанет J Редкие Диск
.
5
: 11. Дои:10.1186/1750-1172-5-11. ЧВК 2893117. PMID 20492708. - Гольдман, Ли; Шафер, Эндрю И. (21 апреля 2015 г.). Goldman-Cecil Medicine: консультации экспертов — онлайн
. Elsevier Health Sciences. ISBN 9780323322850 . - Картон, Джеймс (2012-02-16). Оксфордский справочник по клинической патологии
. ОУП Оксфорд. ISBN 9780191629938 . - Перкин, Рональд М .; Newton, Dale A .; Свифт, Джеймс Д. (2008). Детская госпитальная медицина: Учебник по ведению стационара
. Липпинкотт Уильямс и Уилкинс. ISBN 9780781770323 . - Галанелло, Ренцо; Орига, Рафаэлла (21 мая 2010 г.). «Бета-талассемия». Журнал редких заболеваний Orphanet
.
5
(1): 11. Дои:10.1186/1750-1172-5-11. ISSN 1750-1172. ЧВК 2893117. PMID 20492708. - Введение в патологию для ассистента физиотерапевта
. Издательство «Джонс и Бартлетт». 2011 г. ISBN 9780763799083 . - Андерсон, Грегори Дж .; Макларен, Гордон Д. (16 января 2012 г.). Физиология и патофизиология железа у людей
. Springer Science & Business Media. ISBN 9781603274845 . - Бартон, Джеймс С.; Эдвардс, Корвин Q .; Phatak, Pradyumna D .; Бриттон, Роберт С .; Бэкон, Брюс Р. (22.07.2010). Справочник по расстройствам, связанным с перегрузкой железом
. Издательство Кембриджского университета. ISBN 9781139489393 . - McCance, Kathryn L .; Хютер, Сью Э. (13 декабря 2013 г.). Патофизиология: биологическая основа болезней взрослых и детей
. Elsevier Health Sciences. ISBN 9780323088541 . - Леонард, Дебра Г. Б. (25 ноября 2007 г.). Молекулярная патология в клинической практике
. Springer Science & Business Media. ISBN 9780387332277 . - Bowen, Juan M .; Маззаферри, Эрнест Л. (2012-12-06). Современная внутренняя медицина: клинические примеры
. Springer Science & Business Media. ISBN 9781461567134 . - Заболевания, Национальная организация редких заболеваний (2003 г.). Руководство NORD по редким заболеваниям
. Липпинкотт Уильямс и Уилкинс. ISBN 9780781730631 . - Бартон, Джеймс С.; Эдвардс, Корвин К. (13 января 2000 г.). Гемохроматоз: генетика, патофизиология, диагностика и лечение
. Издательство Кембриджского университета. ISBN 9780521593809 . - Уилкинс, Липпинкотт Уильямс и (2009). Профессиональное руководство по болезням
. Липпинкотт Уильямс и Уилкинс. п.513. ISBN 9780781778992 . большая талассемия. - Уорд, Аманда Дж; Купер, Томас А (2009). «Патобиология сращивания». Журнал патологии
.
220
(2): 152–63. Дои:10.1002 / путь.2649. ЧВК 2855871. PMID 19918805. - «определение ДНК». Dictionary.com
. Получено 2015-05-26. - Окпала, Ихэаньи (15 апреля 2008 г.). Практическое ведение гемоглобинопатий
. Джон Вили и сыновья. ISBN 9781405140201 . - Васудеван, Д. М .; Sreekumari, S .; Вайдьянатан, Каннан (01.11.2011). Учебник биохимии для студентов-стоматологов
. JP Medical Ltd. ISBN 9789350254882 . - Taeusch, H. William; Ballard, Roberta A .; Глисон, Кристин А .; Эйвери, Мэри Эллен (2005). Болезни Эйвери новорожденных
. Elsevier Health Sciences. ISBN 978-0721693477 . - Бета-талассемия: новые идеи для медицинских работников: издание 2013 г.: ScholarlyBrief
. Научные издания. 2013-07-22. ISBN 9781481663472 . - «Факторы риска». Клиника Майо
. Получено 4 апреля 2021. - «Как диагностируют талассемии? — NHLBI, NIH». www.nhlbi.nih.gov
. Получено 2015-05-26. - Клетки-мишени, Имперский колледж Лондонского медицинского факультета
- ^ аб
Оркин, Стюарт Х .; Натан, Дэвид Дж .; Гинзбург, Дэвид; Смотри, А. Томас; Фишер, Дэвид Э .; Люкс, Сэмюэл (2009).
Гематология младенчества и детства Натана и Оски
(7-е изд.). Филадельфия: Сондерс. ISBN 978-1-4160-3430-8 .[
страница нужна
] - «Каковы признаки и симптомы талассемии? — NHLBI, NIH». www.nhlbi.nih.gov
. Получено 2015-05-26. - Галанелло, Ренцо; Орига, Рафаэлла (2010). «Бета-талассемия». Журнал редких заболеваний Orphanet
.
5
(1): 11. Дои:10.1186/1750-1172-5-11. ЧВК 2893117. PMID 20492708. - Шрайвер, Ирис (09.09.2011). Диагностическая молекулярная патология на практике: индивидуальный подход
. Springer Science & Business Media. ISBN 9783642196775 . - Cousens, N.E .; Gaff, C.L .; Metcalfe, S.A .; Делатицкий, М. Б. (2010). «Скрининг носителей бета-талассемии: обзор международной практики». Европейский журнал генетики человека
.
18
(10): 1077–83. Дои:10.1038 / ejhg.2010.90. ЧВК 2987452. PMID 20571509. - «Скрининг на признак бета-талассемии: опасности среди населения западноафриканского происхождения». Получено 4 апреля 2021.
- Muncie, Herbert L .; Кэмпбелл, Джеймс С. (2009). «Альфа и бета талассемия». Американский семейный врач
.
80
(4): 339–44. PMID 19678601. - Грир, Джон П .; Arber, Daniel A .; Глэдер, Бертил; Лист, Алан Ф .; Значит, Роберт Т .; Параскевас, Фриксос; Роджерс, Джордж М. (29 августа 2013 г.). Клиническая гематология Винтроба
. Липпинкотт Уильямс и Уилкинс. ISBN 9781469846224 . - Грир, Джон П .; Arber, Daniel A .; Глэдер, Бертил; Лист, Алан Ф .; Значит, Роберт Т .; Параскевас, Фриксос; Роджерс, Джордж М. (29 августа 2013 г.). Клиническая гематология Винтроба
. Липпинкотт Уильямс и Уилкинс. ISBN 9781469846224 . - Гидроксамовые кислоты: достижения в исследованиях и применении: издание 2011 г.: ScholarlyPaper
. Научные издания. 2012-01-09. ISBN 9781464952081 . - «NCBI — Диагностика блокировки ошибок WWW». pubchem.ncbi.nlm.nih.gov
. Получено 2015-05-26. - «Дефероксамин». ivertox.nih.gov
. Получено 2015-05-26. - Саблофф, Митчелл; Чанди, Маммен; Ван, Чживэй; Логан, Брент Р .; Гавамзаде, Ардешир; Ли, Чи-Конг; Ирфан, Сайед Мохаммад; Bredeson, Christopher N .; Коуэн, Мортон Дж. (2011). «Трансплантация HLA-совместимого костного мозга брату и сестре при большой β-талассемии». Кровь
.
117
(5): 1745–1750. Дои:10.1182 / blood-2010-09-306829. ISSN 0006-4971. ЧВК 3056598. PMID 21119108. - «Генная терапия обещает лечить бета-талассемию и серповидно-клеточную анемию». 2012-03-28. Получено 2015-10-15.
- Уранюс, Селман. «Спленэктомия при гематологических нарушениях». NCBI
. Получено 4 апреля 2021. - А, Коэн. «Терапия переливания крови при большой β-талассемии». NCBI
. Получено 4 апреля 2021. - CRISPR Therapeutics и Vertex Pharmaceuticals принимают меры для начала первого клинического испытания CRISPR / Cas9 в Европе в 2018 году. Автор Клара Родригес Фернандес, 13/12/2017
- Каппеллини, Мария Доменика (2007). «Exjade® (деферасирокс, ICL670) в лечении хронической перегрузки железом, связанной с переливанием крови». Терапия и управление клиническими рисками
.
3
(2): 291–299. Дои:10.2147 / tcrm.2007.3.2.291. ISSN 1176-6336. ЧВК 1936310. PMID 18360637. - Адвани, Пуджа. «Лекарство от бета-талассемии». Medscape
. Получено 4 апреля 2021. - Шварц, М. Уильям (2012). 5-минутная педиатрическая консультация
. Липпинкотт Уильямс и Уилкинс. ISBN 9781451116564 . - Порвит, Анна; Маккалоу, Джеффри; Эрбер, Венди Н. (27 мая 2011 г.). Патология крови и костного мозга
. Elsevier Health Sciences. ISBN 978-0702045356 . - Гемоглобинопатии
. Издательство Jaypee Brothers. 2006 г. ISBN 9788180616693 . - Торре, Дарио М .; Лэмб, Джеффри С .; Руисвик, Джером Ван; Шапира, Ральф М. (2009). Клиническая медицина Кочара для студентов
. Липпинкотт Уильямс и Уилкинс. ISBN 9780781766999 . - Бриссо, Пьер; Каппеллини, Мария Доменика (2014). «БОЛЕЗНЬ ПЕЧЕНИ». Международная федерация талассемии. Цитировать журнал требует | журнал = (помощь)
- «ВОЗ | Глобальная эпидемиология нарушений гемоглобина и производные показатели обслуживания». www.who.int
. Получено 2015-05-26. - Берг, Шери; Биттнер, Эдвард А. (2013-10-16). Обзор медицины интенсивной терапии MGH
. Липпинкотт Уильямс и Уилкинс. ISBN 9781451173680 . - Гематология — это просто
. АвторДом. 2013-02-06. ISBN 9781477246511 . - Абуэльмагд, Ахмед; Агели, Хусейн М. (2013). Базовая генетика: праймер, охватывающий молекулярный состав генетического материала, экспрессию генов и генную инженерию, а также мутации и генетику человека
. Универсальные издатели. ISBN 9781612331928 . - Weatherall, Дэвид Дж (2010). «Глава 47. Талассемии: нарушения синтеза глобина». В Лихтмане, Массачусетс; Киппс, TJ; Seligsohn, U; Каушанский, К; Прчал, Дж. Т. (ред.). Талассемии: нарушения синтеза глобина
.
Гематология Вильямса
(8-е изд.). Компании McGraw-Hill. - «Статистика талассемии». Правильный диагноз
. Получено 4 апреля 2021. - «Талассемия: генетическое заболевание крови, как ожидается, удвоится в следующие несколько десятилетий». ScienceDaily
. Получено 4 апреля 2021.
Причины и факторы риска развития заболевания
Талассемия – заболевание наследственное, то есть мутация передается от родителя ребенку. Если кто-то из родственников страдал от этой патологии, риск развития заболевания повышается.
Второй фактор риска – этническая принадлежность. Больше всего талассемия распространена в Африке, Средней Азии и странах Средиземноморья, где она и была открыта (в переводе с греческого “талассемия” — это “морская анемия”). Различают два основных типа талассемии: альфа и бета. В первом случае мутация затрагивает гены, отвечающие за синтез альфа-глобинов, во втором – бета.
Альфа-глобины кодируются четырьмя генами. Тяжесть заболевания будет зависеть от количества патологически измененных участков ДНК:
- 1 измененный ген – бессимптомная форма. Но при ней человек становится носителем заболевания и может передать его своим детям;
- 2 гена – легкое течение болезни;
- 3 гена – тяжелое течение болезни;
- 4 гена – редкий тип заболевания, который плохо совместим с жизнью. Большинство плодов гибнет еще в период внутриутробного развития, а родившиеся малыши, в основном, умирают вскоре после рождения или нуждаются в пожизненной терапии. В отдельных случаях удается их вылечить путем пересадки костного мозга.
Бета-глобины кодируются одним геном, который локализуется в 11-й хромосоме. Если дефектный ген содержится только в одной хромосоме из пары, заболевание протекает легко (малая талассемия). Повреждение обеих хромосом приводит к очень серьезному заболеванию, известному как большая талассемия или болезнь Кули (анемия Кули).
Содержание
- 1 Признаки и симптомы
- 2 Причина 2.1 Мутации
- 2.2 сборка мРНК
- 4.1 Анализ ДНК
- 6.1 Большая бета-талассемия 6.1.1 Хирургический
- 7.1 Эволюционная адаптация
Симптомы и признаки талассемии
В большинстве случаев талассемию определяют еще на этапе дородовой диагностики. При необходимости лечение начинают сразу, не дожидаясь появления симптомов. Если заболевание не выявила пренатальная диагностика, ожидаются следующие симптомы:
- бледность или желтушность слизистых оболочек;
- замедленный рост;
- темная моча;
- увеличение живота;
- деформация костей, особенно костей черепа.
Время появления первых признаков талассемии во многом зависит от типа заболевания и количества мутаций. У одних детей симптомы регистрируются вскоре после рождения, у других – в первые два года жизни.
Диагностика талассемии
Симптомы при талассемии бывают более или менее характерными. Чтобы поставить окончательный диагноз, врачу необходимы результаты лабораторных исследований. Обязателен при подозрении на талассемию общий анализ крови. Он покажет сниженное количество эритроцитов мелких, светлых, разных по форме и размеру. Кроме зрелых клеток, в мазке будет немало их предшественников — бластов. Дополнительно могут назначить другие специфические анализы крови для определения степени тяжести нарушений (биохимический анализ, определение железосвязывающей способности плазмы или ферритина в сыворотке). Также разработаны молекулярные тесты (ПЦР), позволяющие определить наличие мутаций.
Для оценки состояния печени, селезенки используют УЗИ, а для выявления патологии костной ткани – рентгенографию.
У ребенка талассемия может быть диагностирована еще на этапе вынашивания. Это исследование особенно рекомендуется проводить родителям, которые больны или могут быть носителями этого заболевания. Существует два метода диагностики:
- биопсия ворсинок хориона – проводится на 11-ой неделе беременности;
- амниоцентез (отбор околоплодных вод) – назначают на 16-й неделе.
Профилактика
Бета-талассемия — это наследственное заболевание, позволяющее проводить профилактическое лечение путем скрининга на носительство и пренатальной диагностики. Это можно предотвратить, если у одного из родителей есть нормальные гены, что дает основания для скрининга, позволяющего носителям выбирать партнеров с нормальным гемоглобином. Исследование направлено на выявление генов, которые могут дать потомство с серповидно-клеточной анемией. Пациенты с диагнозом бета-талассемия имеют MCH ≤ 26 пг и RDW [31] Эта процедура скрининга оказалась нечувствительной в популяциях западноафриканского происхождения, поскольку по показателям высока распространенность альфа-талассемии. В странах есть программы распространения информации о репродуктивных рисках, связанных с носителями гемоглобинопатий. Программы скрининга носителей талассемии включают образовательные программы в школах, вооруженных силах и в средствах массовой информации, а также консультирование носителей и пары носителей.[32] Скрининг показал снижение заболеваемости; к 1995 г. распространенность в Италии снизилась с 1: 250 до 1: 4000, а в этом регионе — на 95%. Снижение заболеваемости принесло пользу больным талассемией, поскольку потребность в крови снизилась, что улучшило предложение лечения.[нужна цитата
]
Лечение талассемии
Определяется типом и степенью тяжести. При умеренно выраженных симптомах лечение не назначают. Время от времени проводят только переливание крови. В основном это нужно после операций, родов или для предотвращения возможных осложнений. Люди с бета-талассемией требуют более частых переливаний крови. Для нормализации избыточного уровня железа им также назначают специфические препараты, которые связывают и выводят железо.
При выраженной и тяжелой формах болезни существует два способа лечения:
- частые переливания крови (раз в несколько недель), которые сочетают с приемом препаратов, выводящих лишнее железо из организма;
- пересадка костного мозга – единственный метод, который помогает полностью излечить человека от талассемии. К сожалению, далеко не всегда трансплантация бывает успешной.
Лекарственные препараты при талассемии назначают только для коррекции симптомов и осложнений. Медикаментозной терапии самого заболевания не существует.
дальнейшее чтение
- Цао, Антонио; Галанелло, Ренцо (2010). «Бета-талассемия». В Пагоне, Роберта А; Bird, Thomas D; Долан, Синтия Р.; Стивенс, Карен; Адам, Маргарет П. (ред.). GeneReviews
. Вашингтонский университет, Сиэтл. PMID 20301599. - Бахал, Раман; Макнер, Николь Али; Кихано, Элиас; Лю, Яньфэн; Сулковски, Паркер; Турчик, Одри; Лу, И-Цзянь; Bhunia, Dinesh C .; Манна, Арунава; Greiner, Dale L .; Brehm, Michael A .; Ченг, Кристофер Дж .; Лопес-Хиральдес, Франсеск; Риккарди, Адель; Белоор, Джагадиш; Краузе, Дайан С .; Кумар, Прити; Галлахер, Патрик Дж .; Брэддок, Деметриос Т .; Зальцман, В. Марк; Ly, Danith H .; Глейзер, Питер М. (26 октября 2021 г.). «Коррекция in vivo анемии у β-талассемических мышей с помощью γPNA-опосредованного редактирования генов с доставкой наночастиц». Nature Communications
.
7
: 13304. Bibcode:2016НатКо … 713304B. Дои:10.1038 / ncomms13304. ISSN 2041-1723. ЧВК 5095181. PMID 27782131.
Осложнения талассемии
Возможные осложнения заболевания:
- излишек железа, которое входит в состав гемоглобина. Накопление этого элемента в организме приводит к поражению сердца, печени, эндокринной системы;
- подверженность инфекциям. Особенно актуально для пациентов, которым удалили селезенку;
- деформация костей, связанная с увеличением объема костного мозга. Чаще всего этот процесс затрагивает кости черепа, реже конечностей. Они истончаются и чаще ломаются;
- спленомегалия — увеличение селезенки, где в основном и гибнут дефектные эритроциты. Если селезенка увеличена очень сильно, ее удаляют. Эта операция называется спленэктомия;
- задержка роста и полового созревания;
- сердечные болезни (хроническая сердечная недостаточность и аритмии) могут развиться при тяжелом течении заболевания.
Сколько стоит «Зинтегло»?
P. S. «Блюбёрд» выставила стоимость «Зинтегло» в размере 1,575 млн евро (1,77 млн долларов). Как и предполагалось, оплата будет осуществляться в рассрочку на протяжении пяти лет равными долями: вначале аванс, а затем ежегодные платежи, но только в том случае, если зафиксирована результативность лечения.
«Блюбёрд» еще не определилась с конечной стоимостью своей генотерапии, но она, очевидно, окажется запредельной. Понятное дело, в высокоразвитых странах со страховой медициной никто из пациентов не платит всю сумму из собственного кармана.
Да, по подсчетам «Блюбёрд», «справедливая цена» «Зинтегло», рассчитанная с учетом улучшения качества и продолжительности жизни пациента, находится в пределах 2,1 млн долларов, но, как ожидается, итоговая стоимость будет существенно меньше. Указанная астрономическая сумма исходит, очевидно, из тех совокупных затрат системы здравоохранения на многолетнюю хроническую терапию бета-талассемии. Так, по оценкам экспертов, один американский пациент с этим заболеванием обходится где-то в 200–300 тыс. долларов ежегодно.
Предложено в первый год внести 20% авансом, а оставшуюся сумму выплачивать 20-процентным частями в рассрочку ежегодно в течение четырех лет, причем при условии, что эффективность лечения удовлетворяет предопределенным критериям, таким как исчезновение необходимости в переливаниях крови или существенное снижение числа таких процедур.
Другими словами, платить придется лишь по факту результативности генотерапии, тем более пока нет неоспоримых данных о продолжительности сохранения ее целительного действия. Подход кажется разумным, если принимать во внимание различия в промежуточных исходах среди больных, хотя общая эффективность «Зинтегло» и правда впечатляет.
Согласно прогнозам отраслевых экспертов, стоимость «Зинтегло» составит 1,2 млн долларов в США и 900 тыс. долларов (785 тыс. евро) в Европе.
Проблемы терапии
Лечение талассемии зависит от тяжести поражения эритропоэза, степени охвата генов процессами мутации. В настоящее время применяются следующие методы.
Диетический рацион направлен на снижение всасывания железа в кишечнике, рекомендуются орехи, какао, соя, чай.
Тяжелая форма требует регулярного переливания крови, эритроцитарной массы, размороженных и профильтрованных эритроцитов. Эффективность временная, возможны побочные эффекты, но главное — сохранить жизнь больному.
Терапия дополняется ежедневным устранением излишков железа с помощью введения (хелатов — специальных комплексов, повышающих действие лекарства). Для связывания железа назначается Десферал. Этот препарат предупреждает сидероз (патологическое состояние, вызванное отложением в тканях железа), но на уровень гемоглобина не влияет.
При возникновении резкого ухудшения состояния по аналогии с гемолитическими кризами показаны глюкокортикоиды в больших дозах.
Спленэктомия возможна при больших размерах селезенки детям после пятилетнего возраста. Наиболее оптимален возраст 8–10 лет. После удаления селезенки наступает период улучшения, но опасен риск присоединения инфекции.
Для трансплантации необходим донор, совпадающий по всем параметрам, лучше всего из близких родственников.
К симптоматическим средствам относятся препараты гепатопротекторного действия, большие дозы аскорбинки помогают вывести излишки железа из организма.
Все формы талассемии нуждаются в препаратах с фолиевой кислотой и витаминами группы В. На фоне присоединившейся инфекции, при беременности следует применять фолиевую кислоту в больших дозах, поскольку неэффективное кроветворения при талассемии значительно увеличивает ее потребление клетками.