24 марта 2021
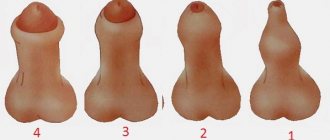
Размер яичек – один из показателей здоровья половых органов, поэтому любые изменения как в большую, так и в меньшую сторону должны насторожить. В нормальном состоянии яички имеют длину 4-6 см и ширину в пределах 2-3,5 см. Уменьшение размеров встречается редко, но может происходить под воздействием внешних факторов или ряда заболеваний. Причина, почему у мужчины маленькие яички, может быть как врожденной, так и приобретенной. Перечислим все возможные варианты возникновения такого состояния.
Гипоплазия яичек — что это такое
Заболевание встречается у 5-6% новорожденных мальчиков и бывает как односторонним, так и двусторонним. Степень гипоплазии может быть различна: от незначительного уменьшения яичка в размерах до полного отсутствия гонад. В рудиментарных текстикулах извитые канальцы практически отсутствуют, недоразвитые яички нередко даже не опускаются в мошонку.
При односторонней гипоплазии яичек концентрация гормонов в организме снижена незначительно, так как второй орган пребирает на себя функции первого, увеличиваясь в размере. При двусторонней форме наблюдается недостаток тестостерона и отсутствие фертильности. Таким образом, при данной патологии отмечается не только уменьшение органа в размерах, нарушается образование спермы и выработка мужских половых гормонов.
Варикоцеле
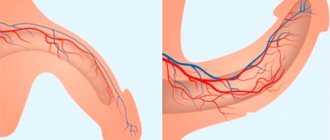
Сосуды, обеспечивающие кровоснабжение яичек, имеют большое число сообщений между собой, образующих лозовидное сплетение. По некоторым причинам кровеносная сетка расширяется. Одной из распространенных причин, почему яички становятся маленькими, выступает варикоцеле. Это и есть расширение вен семенного канатика, которое провоцирует избыточный приток крови. Уменьшение размеров составляет около 20%.
Расширение сосудов происходит со стороны расширения вен, в 96% случаев слева. Этим и объясняется, почему одно яичко меньше другого при варикоцеле. Размеры уменьшаются при значительном нарушении кровоснабжения мошонки, когда ее сосудистая система переполняется венозной кровью. В качестве осложнений может нарушаться сперматогенез, развиваться ранний мужской климакс или бесплодие.
На варикоцеле указывают боль в мошонке, неудобство при ходьбе. Возможны усиленное потоотделение, проблемы с половой функцией. Кроме изменения размеров заметно опущение яичка на пораженной стороне, что приводит к его асимметрии и отвисанию. Мошонка становится дряблой, создается ощущение, что она «усыхает». Все потому, что яичко истощается, начинает разрушаться его внутренняя структура. При соответствующем лечении мошонка вырастает до нормальных размеров.
Причины заболевания
Заболевание развивается в результате нарушений в эмбриональном развитии на ранних этапах беременности. Чем раньше произошли повреждения, тем существеннее будут нарушения. Главная причина гипоплазии яичек — это хромосомные и генетические аномалии (повреждение генов, нарушение числа половых хромосом или их структуры). Примером подобного явления может служить синдром Клайнфельтера, Шерешевского-Тернера. Также заболевание может развиваться при гипогонадизме, при гипоплазии других органов эндокринной системы, опухолях надпочечников, алкогольной эмбриофетопатии, гипофизарном нанизме и др.
Существует целый ряд неблагоприятных факторов, которые могут влиять на вероятность развития болезни. Патологии беременности, имеющиеся гормонально активные опухоли, гормональный дисбаланс, повреждение нервной системы ребенка во время родов и др. — эти факторы предрасполагают к развитию заболевания. Кроме того, в некоторых случаях причиной патологии выступает аутоиммунное поражение ткани тестикул. Вероятность возникновения болезни существенно возрастает, если недуг носит семейный характер.
Синдром микропении у детей и подростков: патогенез, клиника, диагностика
Микропения — это аномальное уменьшение длины полового члена на более чем 2,5 SD у здоровых мальчиков, что во взрослом состоянии может вызывать затруднение в полноценной половой жизни. Нарушение формирования полового члена у мальчиков может быть обусловлено структурными или гормональными нарушениями в гипоталамо-гипофизарно-гонадной системе во внутриутробном периоде. Микропенис обычно распознается вскоре после рождения. Термин «микропенис» наиболее часто применяется при отсутствии изменений пениса и мошонки.
Эмбриогенез половой системы
Формирование полового тракта в эмбриогенезе определяется взаимодействием трех групп факторов: генетического механизма, внутренних эпигенетических факторов (ферментные системы, гормоны) и внешних эпигенетических факторов, отражающих влияние внешней среды.
Генетический пол будущего ребенка предопределяется в момент слияния яйцеклетки и сперматозоида и обусловлен набором половых хромосом, образующихся в зиготе при соединении материнской и отцовской гамет (XX — женский, XY — мужской), набором особых генов, определяющих прежде всего тип гонад, уровнем активности ферментных систем, реактивностью тканей к половым гормонам, концентрацией половых гормонов. Генетический мужской пол определяет Y-хромосома (в том числе ген SRY, относящийся к семейству ДНК-регуляторных генов Sox). Ген SRY кодирует регуляторный фактор TDF (Testis-Determining Factor). TDF обуславливает дифференцировку мужского типа гонад из изначально бипотентных половых желез.
Мужские и женские половые железы развиваются из одного недифференцированного зачатка. На 3–4 неделе происходит закладка первичной гонады, формирование парных вольфовых протоков, а затем мюллеровых протоков. До 6-й недели эмбриональной жизни зародыш морфологически одинаков как для женского, так и для мужского пола.
На 6–7 неделе появляются половой бугорок, уретральная щель, ограниченная уретральными и лабиоскротальными складками. Критическая стадия развития индифферентных гонад — 8-я неделя внутриутробного развития. Под влиянием регуляторного фактора TDF, а также генов Sox гонадные валики развиваются как тестикулы из мозгового слоя зародыша.
В настоящее время доказано, что ген, детерминирующий дифференцировку зачатка гонады по мужскому типу, определяет биосинтез специфического мембранного белка, H-Y-антигена. В клетках развивающегося организма, в том числе в клетках, покрывающих поверхность примордиальной гонады, содержатся рецепторы к H-Y-антигену. Захват H-Y-антигена этими клетками индуцирует развитие первичной гонады в яичко [1].
Элементами развивающихся тестикул являются сперматогонии и мезенхимальная ткань. Из клеток мезенхимы у эмбрионов мужского пола образуются клетки Лейдига, которые секретируют тестостерон с 9-й недели под контролем гонадотропинов (хорионического и гипофизарного). Высокая гормональная активность фетальных яичек необходима для дальнейшего формирования полового тракта у плода мужского пола.
Следующим этапом полового формирования становится дифференцировка внутренних и наружных гениталий. На ранних стадиях эмбриогенеза половая система имеет бисексуальные закладки внутренних и наружных половых органов. Внутренние половые органы дифференцируются на 10–12 неделе внутриутробного периода. Основой их развития являются индифферентные вольфовы (мезонефральные) и мюллеровы (парамезонефральные) протоки.
При развитии плода мужского пола мюллеровы протоки регрессируют под влиянием фактора, синтезируемого клетками Сертоли фетальных яичек и названного «антимюллеровым». Вольфовы протоки дифференцируются в придатки яичек, семенные пузырьки, семяпроводы. Формирование половых путей по мужскому типу возможно только при наличии полноценного, активного эмбрионального тестикула.
Наружные половые органы формируются с 12-й по 20-ю неделю внутриутробного периода. Основой развития наружных половых органов плодов обоего пола являются половой бугорок, лабиоскротальные валики и урогенитальный синус.
Урогенитальный синус дифференцируется в предстательную железу и мочеиспускательный канал; урогенитальный бугорок — в половой член и кавернозные тела; половые валики — в мошонку. Маскулинизация наружных гениталий у мужского плода заключается также в атрофии влагалищного отростка урогенитального синуса, срастании мошоночного шва, увеличении кавернозных тел полового члена и формировании мочеиспускательного канала по мужскому типу.
Нисхождение яичек из брюшной полости начинается с 3-го месяца эмбриональной жизни, а к 8–9 месяцу тестикулы опускаются в мошонку. Их опускание обусловлено как механическими факторами (внутрибрюшное давление, атрофия и укорочение пахового тяжа, неравномерный рост структур, участвующих в этом процессе), так и гормональными (влияние плацентарных гонадотропинов, андрогенов фетальных яичек, гонадотропных гормонов гипофиза плода). Нисхождение яичек совпадает с их максимальной андрогенной активностью [2].
Таким образом, дифференцировка полового тракта происходит в период раннего эмбриогенеза, предопределяется генетическими факторами и обусловливается гормональной функцией фетальных гонад.
Основные звенья гормональной регуляции половой функции организма
Условно можно выделить три основных уровня гормональной регуляции: а) центральный — кора головного мозга, подкорковые образования, ядра гипоталамуса, эпифиз, аденогипофиз; б) периферический — половые железы, надпочечники и секретируемые ими гормоны и их метаболиты; в) тканевый — специфические рецепторы в органах-мишенях, с которыми взаимодействуют половые гормоны и их активные метаболиты.
Координирующим звеном гормональной регуляции являются подкорковые образования и гипоталамус, которые осуществляют взаимосвязь между центральной нервной системой, с одной стороны, и гипофизом и половыми железами, с другой. В ядрах гипоталамуса найдено высокое содержание биогенных аминов и нейропептидов, играющих роль нейротрансмиттеров и нейромодуляторов в трансформации нервного импульса в гуморальный. Ядра миндалины оказывают как стимулирующее, так и ингибирующее воздействие на гонадотропную функцию гипофиза, что зависит от локализации импульса.
Из веществ, обнаруженных в эпифизе, наиболее изученными в регуляции гонадотропной функции представляются индольные соединения — мелатонин и серотонин. Ингибирующее влияние мелатонина на антигонадотропную функцию реализуется на уровне гипоталамуса, блокируя синтез и секрецию гонадотропинов рилизинг-гормона (ЛГ-РГ). Кроме того, в эпифизе обнаружены и другие вещества пептидной природы с выраженным антигонадотропным действием, превышающим активность мелатонина в 60–70 раз [3].
В ядрах среднего и отчасти заднего гипоталамуса образуются рилизинг-гормоны — вещества, регулирующие все тропные функции аденогипофиза. Гипоталамус осуществляет регуляцию половой (гонадотропной) функции посредством синтеза и секреции гонадотропинов ЛГ-РГ.
Действие ЛГ-РГ стимулирует выброс лютеинизирующего гормона (ЛГ) и фолликулостимулирующего гормона (ФСГ). В гипоталамусе выделяют центры, осуществляющие тоническую и циклическую секрецию гонадотропинов. Тонический центр секреции ЛГ-РГ обеспечивает постоянное выделение гонадотропных гормонов. Присутствие андрогенов является необходимым условием для развития регуляции по мужскому типу.
Непосредственно в регуляции половой системы принимают участие три тропных гормона гипофиза: ЛГ, ФСГ и пролактин (ПРЛ). Другие гипофизарные гормоны — тиреотропный (ТТГ), соматотропный (СТГ), адренокортикотропный (АКТГ) — также влияют на стероидпродуцирующие клетки гонад.
ЛГ является основным гормоном, обеспечивающим синтез основного андрогена — тестостерона, который вырабатывается в интерстициальных клетках Лейдига тестикулов. Последний в физиологических условиях является основным ингибитором секреции ЛГ. Большие дозы пролактина уменьшают количество рецепторов к ЛГ [4].
Индукция стероидогенеза в клетках Лейдига под влиянием ЛГ происходит при активации фермента 20a-гидроксилазы, обеспечивающего переход холестерина в прегненолон. Основным андрогеном в мужском организме является тестостерон. Кроме тестостерона, в клетках Лейдига вырабатываются андрогены с меньшей биологической активностью: дегидроэпиандростерон и Δ4-андростендион. Однако основное количество этих слабых андрогенов образуется в сетчатой зоне надпочечников или служит продуктом периферического превращения тестостерона.
Механизм действия андрогенов на клетку органов-мишеней связан с образованием активного метаболита тестостерона — дигидротестостерона. Тестостерон превращается в активную фракцию непосредственно в клетке под воздействием фермента 5α-редуктазы [2].
Гормональная регуляция половой сферы ребенка на основных этапах развития
Гонадостат функционирует на протяжении всего развития ребенка, начиная с внутриутробного периода. В процессе становления половой функции у ребенка можно выделить 4 основных «критических» периода: 1) внутриутробный (фетальный); 2) период новорожденности; 3) препубертатный; 4) пубертатный.
В фетальный период максимальная концентрация тестостерона в крови плода мужского пола, свойственная взрослым мужчинам, выявляется между 10-й и 18-й неделями внутриутробного развития. Это обусловлено прежде всего участием тестостерона и его активного метаболита 5α-дигидротестостерона в формировании внутренних и наружных гениталий мальчика. Рост генитального бугорка продолжается в течение всего пренатального периода.
На первом году жизни происходит резкое снижение уровня половых гормонов. Качественные изменения гормональной сферы наступают после 6 лет, когда происходит созревание адреналовой системы с быстрым подъемом уровня надпочечниковых андрогенов: дегидроэпиандростерона (ДГЭА) и его сульфата (ДГЭА-С) и Δ4-андростендиона.
Важнейшим этапом полового развития ребенка является пубертат. В этот период происходит сложная многоэтапная перестройка гипоталамо-гипофизарных взаимоотношений. По мере увеличения тестикул увеличиваются размеры пениса: вначале длина, а затем его диаметр [5].
Итак, формирование наружных гениталий у мальчиков в эмбриональный период требует значительного количества андрогенных гормонов. Снижение биосинтеза тестостерона в эмбриональном тестикуле или дефект 5a-редуктазной активности нарушает маскулинизацию наружных половых органов вплоть до формирования ложного мужского гермафродитизма.
Клиника микропении
Диагноз микрочлена ставится на основании правильно измеренной длины пениса. Если длина вытянутого полового члена меньше на 2,5 стандартных отклонения, чем у среднестатистического пациента с нормальными внутренними и внешними мужскими половыми органами, ставится диагноз микрочлена. Нормальная длина пениса у доношенного мальчика при рождении 3,9 ± 0,8 см, при этом 1,9 см составляет крайнюю допустимую величину в пределах 2,5 SD. К началу пубертата — соответственно 6,4 ± 1,1 см, 3,7 см, у взрослого мужчины — соответственно 13,3 ± 1,6 см, 9,3 см [2, 6].
Мошонка присутствует, хотя в некоторых случаях может быть недоразвита. Тестикулы находятся в мошонке, но функция их может быть снижена. У некоторых пациентов нарушается опускание яичек. Объем тестикул ниже нормальных значений. Микропения является симптомом многих врожденных эндокринных аномалий, протекающих с гипер- или гипогонадотропным гипогонадизмом [7].
Синдром Кляйнфельтера
Синдром Кляйнфельтера (СК) — патология половой хромосомы, проявляется наличием как минимум одной лишней X хромосомы (47ХХУ) или мозаичным кариотипом. Встречается с частотой 1:300–1000.
До пубертатного возраста у мальчиков с СК могут обнаруживаться крипторхизм и маленькие размеры полового члена. В пубертатном периоде характерна гинекомастия, высокорослость, евнухоидные пропорции тела. Размер тестикул остается допубертатным, консистенция их плотная. Данный синдром характеризуется кариотипом 47XXY, гипергонадотропным гипогонадизмом (гипоплазия яичек и микропения), бесплодием, сниженным количеством волос на теле, гинекомастией, снижением интеллекта, деформацией костей и высоким ростом.
Наличие в мужском кариотипе дополнительной Х-хромосомы не влияет на дифференцировку тестикул и формирование гениталий по мужскому типу. Однако жизнедеятельность герминативных клеток нарушается, сперматогенез отсутствует. Это приводит у взрослых больных к азооспермии и бесплодию [8].
ХХ-мужчины
Частота встречаемости кариотипа ХХ у мальчиков составляет примерно 1:20 000. Причина заболевания не во всех случаях доказана. У части пациентов выявляют транслокацию SRY отцовской Y-хромосомы на Х-хромосому в период мейоза. Фенотипически они соответствуют мужскому полу. Половой член и мошонка сформированы по мужскому типу, однако половой член может быть укорочен, встречается гипоспадия. Внутренние половые органы соответствуют мужскому полу. Тестикулы в пубертатном возрасте остаются небольшого размера, плотные, как у пациентов с синдромом Кляйнфельтера. Взрослые ХХ-мужчины способны к половой жизни, но они бесплодны. Высокорослость для них не характерна, интеллект не нарушен. Содержание ЛГ и ФСГ повышено.
Синдром Нунан
В 50% возможна мутация в гене PTPN11 (12q24.1), у 25% мутации в SOS гене (2р22-р21). Проявляется крипторхизмом, микропенисом и гипоплазией мошонки. Помимо этого у части больных в пубертатном возрасте формируется клиника евнухоидизма. Нередко при этом синдроме выявляются крыловидные складки на шее, треугольное лицо, вальгусная деформация локтевых суставов, низкорослость, лимфатические отеки кистей и стоп, птоз, впалая грудная клетка, пороки правых отделов сердца, умственная отсталость. Низкий уровень тестостерона в крови сочетается с повышением уровней ЛГ и ФСГ.
Синдром анорхизма
Синдром анорхизма это внутриутробное, генетически детерминированное поражение гонад (мутации гена INSL на 19р). При рождении наружные половые органы сформированы правильно, но в некоторых случаях встречается микропенис и уменьшение мошонки. Яички отсутствуют. Экстрагенитальные признаки гипогонадизма формируются после 12–14 лет. Половое созревание в пубертатном периоде отсутствует. Наблюдается инволюция вторичных половых признаков, ожирение и евнухоидизм. В крови низкий уровень тестостерона, но высокое содержание ЛГ и ФСГ. Проба с хорионическим гонадотропином человека (ХГЧ) отрицательная.
Дисгенезия гонад
Причиной недоразвития фетального яичка могут быть количественные и структурные хромосомные аберрации, не исключена генная мутация. При кариотипе 46ХY возможны структурные аномалии Y-хромосомы, захватывающие область SRY. Дисгенетичные яички не способны выделять в достаточном количестве активный антимюллеровый фактор, а андрогены не обеспечивают регресс мюллеровых протоков и нормальную маскулинизацию наружных гениталий в эмбриональном периоде, что вызывает неправильное формирование как внутренних, так и наружных половых органов. Наружные гениталии имеют более или менее незавершенную эмбриональную маскулинизацию. Степень маскулинизации урогенитального синуса, а также размеры полового члена ребенка могут служить своеобразным критерием в прогнозировании функциональной (андрогенной) активности дисгенетичных тестикул. Клиническая симптоматика: больной имеет дисгенетичное яичко, производные парамезонефральных (влагалище, матку, маточные трубы) протоков и мезонефральных (придаток яичка) протоков. Наружные гениталии имеют бисексуальное строение. Неправильное строение наружных половых органов выявляется при рождении ребенка
Изолированный гипогонадотропный гипогонадизм
При этом синдроме снижена секреция ЛГ и ФСГ в отдельности или оба гормона одновременно. Наследуется аутосомно-рецессивно. Чаще обусловлен блокирующими мутациями гена рецептора гонадотропин-рилизинг-гормона, дефектами гена b-субъединицы ЛГ или ФСГ соответственно. Причиной нарушения секреции гонадотропинов является целюллярная недостаточность аденогипофиза или гипоталамуса. При рождении у мальчика отмечается недоразвитие мошонки, одно- или двусторонний крипторхизм с паховой дистопией гипоплазированных яичек, микропенис. До пубертата формируются евнухоидизм, отсутствие полового созревания, высокий рост, ожирение, гинекомастия. Снижены уровни ЛГ, ФСГ и тестостерона в крови [7].
Вариантом вышеописанного синдрома является врожденный изолированный дефицит ЛТ (Паскуалини синдром). Из-за дефицита ЛГ нарушается выработка в клетках Лейдига андрогенов. Базальный и стимулированный уровень ЛГ снижен, а уровень ФСГ нормальный [3].
Синдром Каллмана
Синдром Каллмана — это генетически разнородная наследственная болезнь, проявляющаяся гипофизарным гипогонадизмом в сочетании с аносмией. Распространенность болезни у мальчиков составляет примерно 1:8000.
Развивается вследствие нарушения закладки и миграции ЛГ-РГ-нейронов и ольфакторных нервов в период органогенеза, приводящего к аносмии и гипогонадизму. Микропенис встречается при грубом дефекте секреции ЛГ-РГ-гормонов. В некоторых случаях могут присутствовать другие аномалии развития, которые включают почечный агенез, заячью губу и/или волчье небо, селективный агенез зубов и двуручную синкинезию [9].
Синдром Прадера–Вилли
Это нейрогенетическое мультисистемное заболевание. Развивается вследствие повреждения критического района хромосомы 15 (сегмента q11.2-q13). Гипогонадизм по гипогонадотропному типу может быть результатом дисфункции гипоталамуса, преимущественно в области вентромедиального и вентролатерального ядер.
В течении заболевания можно выделить две фазы: первая — свойственна детям 12–18 месяцев жизни. Она характеризуется выраженной мышечной гипотонией, снижением рефлексов Моро, сосательного и глотательного, что затрудняет кормление ребенка. Вторая — наступает позже, через несколько недель или месяцев. Проявляется полифагией, постоянным чувством голода, приводящим к развитию ожирения, причем отложение жира наблюдается преимущественно на туловище и в проксимальных отделах конечностей. Мышечная гипотония постепенно уменьшается и к школьному возрасту почти полностью исчезает. Стопы и кисти больных диспропорционально маленькие — акромикрия. Встречаются и другие аномалии: высокий, узкий лоб; миндалевидный разрез глазных щелей с тонкими, опущенными веками; кожа и волосы светлее, чем у всех других членов семьи; гипопигментация радужки; задержка моторного развития; микродонтия, гипоплазия хрящей ушных раковин, сколиоз, глаукома. Отмечается гипогонадизм (гипоплазия полового члена и мошонки, крипторхизм). Рост больных нередко снижен [10].
Синдром Барде–Бидля
Синдром Барде–Бидля — генетическая патология, связанная с цилиопатией. Встречается с частотой 1:120 000. Тип наследования аутосомно-рецессивный. Выделяют несколько генетических форм: BBS1 картирован 11q; BBS2 — 16q21; BBS3 — 3p [3]. Заболевание характеризуется ожирением, микрогенитализмом и крипторхизмом, олигофренией, полидактилией, поликистозом почек, спастической параплегией, пигментной ретинопатией. Уровень ЛГ и ФСГ низкий.
Гиперпролактинемический гипогонадизм
У мальчиков в препубертатном периоде развиваются евнухоидные пропорции тела: относительно длинные конечности, высокая талия, бедра относительно шире пояса нижних конечностей, отложение жира в области сосков, живота, у гребешков подвздошных костей, мышцы дряблые, слабые, голос высокий детский. Тестикулы и пенис могут быть умеренно гипоплазированы. В пубертатном периоде могут отсутствовать вторичные половые признаки. Половое влечение и фертильность снижены.
При гиперпролактинемии ингибируется секреция гонадолиберина и уменьшается частота и амплитуда пульсовой секреции ЛГ, снижается уровень тестостерона в сыворотке крови. В результате блокады 5-альфа-редуктазы происходит снижение конверсии тестостерона в дегидротестостерон, что приводит к развитию клинических признаков гипогонадизма [4].
Недостаточность 3β-гидроксистероиддегидрогеназы (3β-ГСД)
Это одна из редких форм врожденной гиперплазии коры надпочечников, которая проявляется мужским псевдогермафродитизмом. Данный фермент участвует в биосинтезе минералокортикоидов, глюкокортикоидов и половых стероидов. Недостаточная продукция всех физиологически активных стероидов коры надпочечников связана с дефектом гена HSD3B2 1p13.1. Это может приводить к гермафродитному строению наружных гениталий у мальчиков. При гормональном исследовании обнаруживаются снижение уровня 17a-гидроксипрогестерона, кортизола, альдостерона, тестостерона. Перед ферментным дефектом накапливаются Δ5-ненасыщенные стероиды, в том числе большое количество не активного андрогена ДГЭА. Типично повышение уровня АКТГ [11].
У мальчиков недостаточная андрогеновая активность ДГЭА нарушает образование кавернозных тел полового члена, в связи с чем он выглядит недоразвитым. В пубертатном периоде андрогенизация значительно возрастает за счет периферической конверсии ДГЭА в тестостерон, но не достигает уровня взрослых мужчин. Последним необходима поддерживающая терапия препаратами тестостерона.
Недостаточность 17α-гидроксилазы
Недостаточность 17α-гидроксилазы вызывается дефектом гена CYP21 6p21/3 [1]. Мутации гена CYP17 могут клинически проявляться в виде недостаточности 17a-гидроксилазы, 17,20-лиазы или их сочетания. Недостаточность 17a-гидроксилазы характеризуется снижением количества, вплоть до полного отсутствия, половых гормонов, синтезируемых как гонадами, так и надпочечниками, при одновременном повышении синтеза минералокортикоидных предшественников. У новорожденных мальчиков с дефектом гена CYP17 наружные гениталии могут иметь феминное строение или слабо андрогенизированы. В пубертатном возрасте характерны клинические симптомы гипергонадотропного гипогонадизма. Заболевание сопровождается артериальной гипертензией различной степени выраженности и гипокалиемией.
При недостаточности 17a-гидроксилазы снижение содержания кортизола стимулирует синтез кортикотропина, и хотя продукция стероидов повышается, она все равно блокируется на этапе 17a-гидроксилазы. Компенсаторно накапливаются 17-дезоксистероиды, в том числе прегненолон, прогестерон, дезоксикортикостерон и кортикостерон. Снижение синтеза андрогенов приводит к гипогонадизму. Вследствие высокой минералокортикоидной активности возникают гипернатриемия и потеря калия, увеличивается объем плазмы крови, развивается артериальная гипертензия. Обычно выявляют гипокалиемию, на фоне которой снижаются количество альдостерона сыворотки крови и активность ренина плазмы. Диагноз подтверждают при выявлении выраженного повышения содержания 11-дезоксикортикостерона и кортикостерона сыворотки. Количество прегненолона и прогестерона слегка повышено. Концентрации в сыворотке крови 17a-гидроксипрегненолона, 17a-гидроксипрогестерона, 11-дезоксикортизола, ДГЭА-сульфата, андростендиона и тестостерона ничтожны или совсем не определяются. Содержание ФСГ и ЛГ — повышено. Возможна пренатальная диагностика: определение содержания стероидов надпочечников в амниотической жидкости.
Недостаточность 17,20-десмолазы (лиазы)
Диагноз этого заболевания зачастую ставят в период полового созревания лицам с мужским генотипом. Они могут воспитываться как девочки и обращаться с жалобами на отсутствие менструаций и гирсутизм, или как мальчики и тогда обращаться с жалобами на гинекомастию и недоразвитие половых органов. У пораженных лиц мужского пола показатели вирилизации, включая клиторомегалию вплоть до микрофаллоса и развитие вторичных мужских половых признаков в период полового созревания, очень напоминают таковые при недостаточности 5a-редуктазы. Все больные бесплодны. Фермент 17b-гидроксистероиддегидрогеназа или 17-кетостероидредуктаза катализируют превращение андростендиона в тестостерон в тестикулах. Мутации гена изофермента 17b-гидроксистероиддегидрогеназы 3-го типа (HSD17B3), расположенного на хромосоме 9q22 [2], приводят к недостаточному синтезу тестостерона. Большинство их представлено нонсенс-мутациями. Существенной корреляции между генотипом и фенотипом не выявляют. Развитие гениталий по промежуточному типу обычно выявляют при рождении. Чаще всего имеются клиторомегалия, сращение губно-мошоночных складок и слепой влагалищный карман. Яички нередко пальпируются в паховых каналах или губно-мошоночных складках, хотя иногда могут быть расположены и в брюшной полости. Как и при других формах мужского псевдогермафродитизма, внутренние отделы мочеполового тракта развиты нормально. Есть придатки яичка, семявыносящие протоки, семенные пузырьки, семявыбрасывающие протоки. Предстательная железа и производные мюллеровых протоков отсутствуют. Характерной лабораторной находкой бывает повышенное соотношение андростендион/тестостерон, образующееся в результате увеличения количества андростендиона и снижения тестостерона. Возможна пренатальная диагностика у потомства пораженных пациентов, если у последних выявлена специфическая мутация.
Дефект 5a-редуктазы
В норме этот фермент восстанавливает тестостерон до 5α-дигидротестостерона (5α-ДГТ) в клетках и, частности, мочеполовом синусе. 5α-ДГТ является более активным метаболитом, играющим основную роль в процессах дифференцировки наружных гениталий у мальчиков. При его отсутствии развиваются нормальные семенные пузырьки и простата, но половой член и тестикулы недоразвиты [11].
При рождении у таких мальчиков наружные гениталии похожи на женские, поэтому большиство этих детей регистрируют и воспитывают как девочек. Во время полового созревания увеличивается продукция тестостерона. Изменяется архитектоника тела по мужскому типу, повышается общее количество костной и мышечной ткани. Гормональный профиль в этом периоде и во взрослом состоянии соответствует генетическому полу, поэтому многие пациенты меняют свой пол на мужской.
Дефект 17-редуктазы
Речь идет об изолированном нарушении синтеза тестостерона. Андростендион не превращается в тестостерон.
Клиническая симптоматика у больных мужского пола включает проявление недостаточности тестостерона в пренатальном (гипоспадия), а в дальнейшем в пубертатном и постпубертатном периодах симптомы гипогонадизма. Однако андростендион может беспрепятственно превращаться в эстрогены. В пубертатном периоде может развиться гинекомастия.
Синдром тестикулярной феминизации (СТФ)
Заболевание, вызванное полным или частичным отсутствием чувствительности тканей к андрогенам, обусловленное нарушением аффинности рецепторов к андрогенам или пострецепторными дефектами. Синдром тестикулярной феминизации является одной из самых частых причин ложного мужского гермафродитизма.
Причина заболевания — мутации гена рецептора андрогенов (AR). Мутации обусловливают резистентность периферических рецепторов к тестостерону и дегидротестостерону. Синдром наследуется по Х-сцепленному рецессивному типу.
В процессе эмбриогенеза гонады дифференцируются как полноценные функционирующие яички. Однако из-за дефекта гена АR ткани больных нечувствительны к тестостерону и дегидротестостерону — гормонам, формирующим мужской фенотип (уретру, простату, половой член и мошонку), но в то же время сохранена чувствительность к эстрогенам. Это приводит к закономерному (феномен автономной феминизации) формированию женского фенотипа без производных мюллеровых протоков (маточных труб, матки и верхней трети влагалища), так как продукция антимюллеровой субстанции клетками Сертоли не нарушена [12].
В зависимости от степени нечувствительности периферических рецепторов к андрогенам различают полную форму (при полной нечувствительности к андрогенам) и неполную форму (когда чувствительность изначально частично сохранена или частично восстанавливается в пубертатном периоде).
Синдром Морриса
Клиническая картина полной формы (синдром Морриса): наружные половые органы женского типа, со слепо замкнутым влагалищем; хорошо развиты молочные железы (гинекомастия), отсутствие матки, маточных труб и простаты, отсутствие соматических аномалий развития, отсутствие лобкового и подмышечного оволосения. Выбор пола женский [2].
Синдром Рейфейнштейна
Неполная форма (синдром Рейфейнштейна) характеризуется вирилизацией наружных половых органов различной степени. Строение наружных гениталий отражает различную степень дефицита андрогенов в период внутриутробного развития. У детей при этой форме выявляется гипоспадия высокой степени с разделенной мошонкой, микропенис, тестикулы находятся в расщепленной мошонке или в паховом канале. При гистологическом исследовании количество клеток Лейдига увеличено, сперматогенез нарушен. Уровень ЛГ и тестостерона повышен [2].
Гипоплазия и агенезия клеток Лейдига
Аутосомно-рецессивное заболевание, при котором нарушаются дифференцировка клеток Лейдига и синтез тестостерона. Встречается с частотой 1:1 000 000.
Выявляется точечная инактивирующая мутация в гене ЛГ-рецептора (2р21) [13]. При рождении наружные гениталии сформированы правильно. Яички уменьшены, возможен крипторхизм. В пубертатном возрасте формируется клиника евнухоидизма с ожирением, недоразвитием наружных половых органов, отсутствием полового оволосения. Уровень ЛГ высокий, снижена концентрация тестостерона и ДГТ, проба с ХГЧ отрицательная. Уровень ФСГ в детстве нормальный, в пубертате снижен.
Микропения также может развиваться в рамках многих генетических пороков развития, не ассоциированных с половыми хромосомами. Иногда микропенис является симптомом врожденного дефицита гормона роста или врожденного гипопитуитаризма. В некоторых случаях причину микропении установить не удается. У таких пациентов не выявляется эндокринной дисфункции.
Размеры полового члена у подростков довольно индивидуальны, поэтому иногда бывает трудно отличить истинную микропению, связанную с гипогонадизмом, от функциональной. При функциональной задержке полового развития, когда наружные половые органы имеют допубертатный вид, у подростков, как правило, отсутствуют другие признаки полового развития: мутация голоса, оволосение на лице, гипертрофия плечевого пояса, активность потовых и сальных желез; наблюдается отставание в росте. Базальная концентрация гонадотропных и половых гормонов соответствует допубертатным значениям. Помогают в диагностике функциональные пробы с ЛР-РГ. В ответ на введение последнего увеличивается выброс ЛГ. Костный возраст у таких подростков отстает от паспортного на 2–3 года.
О ложной микропении (скрытом половом члене) говорят в том случае, когда половой член нормального размера, правильно сформирован, имеет хорошо развитые кавернозные тела и головку, но скрыт окружающими тканями. Такая картина наблюдается при ожирении — половой член скрыт в избыточной подкожно-жировой клетчатке, но хорошо выводится из нее. В пубертатном периоде у некоторых подростков выявляется гормональная дисфункция.
С целью дифференциальной диагностики проводят исследования:
- кариотипа;
- молекулярно-генетические;
- УЗИ органов малого таза;
- компьютерную и магнитно-резонансную томографию черепа;
- костного возраста;
- офтальмологические;
- гормональные;
- функциональные пробы с ЛГ-РГ, с ХГЧ [14].
При врожденной микропении рекомендуется пролонгированные препараты тестостерона, которые дают лучший эффект при введении на первом году жизни. Назначают 3–4 дозы внутримышечных инъекций по 25 мг тестостерона ципионата или энантата один раз в месяц. При отсутствии эффекта пациент должен быть консультирован урологом-андрологом для решения вопроса о реконструктивной пластике [15].
Литература
- Балаболкин М. И., Клебанова Е. М, Креминская В. М. Дифференциальная диагностика и лечение эндокринных заболеваний (рук-во). М.: Медицина, 2002. 752 с.
- Дедов И. И., Семичева Т. В., Петеркова В. А. Половое развитие детей: норма и патология. М., 2002. 232 c.
- Диагностика и лечение эндокринных заболеваний у детей и подростков / Под ред. проф. Н. П. Шабалова. МЕДпресс, 2009. 544 с.
- Журтова И. Б. Синдром гиперпролактинемии у детей и подростков: оптимизация методов диагностики и лечения. Автореф. дис. д.м.н. М., 2012.
- Руководство по детской эндокринологии / Под ред. Ч. Г. Д. Брука, Р.-Л. С. Браун: пер. с англ. под ред. В. А. Петерковой. М.: ГЭОТАР-Медиа, 2009. 352 с.
- Lee P. A., Mazur T., Danish R. et al. Micropenis. I. Criteria, etiologies and classification // The Johns Hopkins medical journal. 1980. 146 (4): 156–163.
- Nihal Hatipoglu, Selim Kurtoglu. Micropenis: Etiology, Diagnosis and Treatment Approaches // J Clin Res Pediatr Endocrinol. 2013, Dec; 5 (4): 217–223.
- Bonomi M., Rochira V., Pasquali D., Balercia G., Jannini E. A., Ferlin A. Klinefelter syndrome (KS): genetics, clinical phenotype and hypogonadism // J Endocrinol Invest. 2021, Sep 19, p. 1–12.
- Dodé C., Hardelin J.-P. Kallmann syndrome // Eur J Hum Genet. 2009, Feb; 17 (2): 139–146.
- Тозлиян Е. В. Синдром Прадера–Вилли в практике педиатра // Практика педиатра. 2014, март, с. 32–39.
- Дедов И. И., Петеркова В. А. Федеральные клинические рекомендации (протоколы) по ведению детей с эндокринными заболеваниями. М.: Практика, 2014. 442 с.
- Zheng Z., Armfield B. A., Cohn M. J. Timing of androgen receptor disruption and estrogen exposure underlies a spectrum of congenital penile anomalies // Proc Natl Acad Sci USA. 2015, Dec 29; 112 (52): E7194–E7203.
- Yang-Feng T. L., Seeburg P. H., Francke U. Human luteinizing hormone-releasing hormone gene (LHRH) is located on the short arm of chromosome 8 (region 8 p11. 2–p21) // Somat Cell Mol Genet. 1986; 12: 95–100.
- Федеральные клинические рекомендации-протоколы по ведению пациентов с врожденной дисфункцией коры надпочечников в детском возрасте от 01.2013, г. Москва. 14 с.
- Nerli R. B., Guntaka A. K., Patne P. B., Hiremath M. B. Penile growth in response to hormone treatment in children with micropenis // Indian J Urol. 2013, Oct-Dec; 29 (4): 288–291.
В. В. Смирнов1, доктор медицинских наук, профессор А. А. Никитин, доктор медицинских наук, профессор
ФГБОУ ВО РНИМУ им. Н. И. Пирогова МЗ РФ, Москва
1 Контактная информация
Симптомы гипоплазии яичек
У некоторых пациентов болезнь протекает бессимптомно и диагностируется лишь при обследовании по поводу какого-либо другого заболевания. Нередко недуг выявляется при обращении к специалистам из-за бесплодия.
Визуально размеры мошонки меньше нормы. Причина заключается в уменьшении размера одной или обеих желез. Как правило, величина яичка при гипоплазии составляет всего 0,5-2,5 см. При одностороннем поражении разница в величине здорового и патологически измененного яичка может быть очень заметной.
Однако выраженность патологических изменений определяется уровнем тестостерона, т.е. основной признак заболевания — нарушение гормонального фона. Нередко при одностороннем поражении сперматогенез протекает нормально; сперма такого мужчины способна оплодотворить яйцеклетку. При двусторонней гипоплазии яичек гормональный фон нарушен всегда, что приводит к недостатку тестостерона и, как следствие, у больного развивается снижение полового влечения, импотенция, при этом вторичные половые признаки слабо выражены. У пациентов старше 14 лет отсутствует или недостаточно выражено подмышечное и лобковое оволосение, наружные половые органы инфантильны, тело непропорционально и напоминает женский тип, голос высокий и др.
Гипотрофия и атрофия
У мужчины бывает маленький размер яичек на фоне приобретенной гипотрофии или атрофии. Ее может вызывать варикоцеле и ряд других проблем или заболеваний:
- перекрут яичка (острое состояние, которое при отсутствии помощи за 6 часов может привести к атрофии из-за прекращения кровоснабжения;
- гидроцеле (водянка яичка, при которой в мошонке скапливается жидкость);
- травмы паховой области, операции на половых органах или по поводу паховой грыжи;
- воспалительные или инфекционные процессы (бактериальный или вирусный орхит, эндемический паротит, инфекции, передающиеся половым путем);
- заместительная терапия тестостероном (длительный прием гормональных препаратов);
- прием анаболических средств и эстрогенов;
- продолжительное воздействие высоких или низких температур на область мошонки;
- нарушение гормонального фона, сбой в работе эндокринной системы;
- злокачественные или доброкачественные образования яичек.
Операции на мошонке и паховом канале могут сопровождаться сдавливанием семенной артерии. Это приводит к нарушению кровоснабжения, которое провоцирует атрофию. К травматическим причинам также относятся повреждения поясничного отделка позвоночника, вызывающие нарушение иннервации яичек.
К факторам риска относятся злоупотребление алкоголем и ожирение, которые способствуют развитию дефицита тестостерона. Первым и самым главным признаком атрофии выступает отмирание тканей. Это и есть причина, почему у мужчины стали меньше яички. Меняется их консистенция, они становятся дряблыми, напоминают мешок без содержимого, хотя половая функция пока сохраняется.
Ввиду уменьшения размеров яичка снижается количество вырабатываемого тестостерона, начинает синтезироваться меньше спермы. Атрофические процессы могут происходить в течение длительного времени, а возникать еще на этапе внутриутробного развития, что проявляется в виде крипторхизма (неопущения яичка в мошонку).
Диагностика
При опущенном в мошонку яичке диагностика гипоплазии яичка не представляет трудностей. Однако эта патология нередко сочетается с крипторхизмом — в этом случае яичко расположено в полости брюшины или в паховом канале. Обнаружить его можно лишь с помощью УЗИ органов мошонки или диагностической лапароскопии. Эти методы позволяют обнаружить не только сам орган, но и выяснить его локализацию, а также провести дифференциальную диагностику с монорхизмом, анорхизмом — полным отсутствием яичек, эктопией (расположение тестикула в противоположной половине мошонки, под кожей в паховой области бедра, в промежности). Под контролем УЗИ выполняется биопсия.
При нарушении либидо, импотенции, избыточном весе проводится исследование уровня тестостерона. С целью диагностики хромосомных аномалий рекомендовано обследование кариотипа и проведение генетического анализа. У взрослых пациентов для оценки параметров эякулята необходимо провести спермограмму — анализ, позволяющий выявить мужской фактор бесплодия. Это исследование необходимо также пройти при подготовке к ЭКО и ИКСИ.
Как влияет нарушение функции яичек на продолжение рода
Уменьшение яичек у мужчин напрямую связано с повышенным риском бесплодия.
Именно в яичках происходит сперматогенез — формирование и развитие мужских половых клеток. Нарушение этого процесса зависит не столько от фактического уменьшения яичек, сколько от сопутствующих изменений структуры и качества тканей репродуктивной сферы.
Клинически значимая гипоплазия практически всегда сочетается с недостатком или отсутствием сперматозоидов, их низкой подвижностью и прочими аномалиями.
Нарушение гормональной функции яичек также неблагоприятно сказывается на детородной функции. Тестостероном регулируется множество сложных процессов, происходящих как в половых органах, так и за их пределами. Даже незначительный дисбаланс выработки и утилизации половых гормонов может приводить к длительному бесплодию.
Как лечить гипоплазию яичка
В большинстве случаев лечение гипоплазии яичка консервативное, во время которого проводится стимуляционная и заместительная терапия. Следует помнить, что препараты, дозировка, продолжительность приема медикаментов назначается только врачом, при этом тактика лечения для каждого пациента подбирается строго индивидуально.
Стимуляция гормонами позволяет увеличить активность сперматозоидов, причем удается добиться лечебного эффекта на довольно продолжительное время. При двусторонней патологии с помощью заместительной терапии в подростковом периоде возможно развить вторичные половые признаки. Кроме того, с помощью гормонотерапии удается существенно увеличить результативность ЭКО и ИКСИ. При необходимости можно воспользоваться криоконсервацией спермы для применения ее в отдаленный период.
Хирургическое лечение проводится при гипоплазии одного яичка, когда второе функционирует нормально. После проведения операции — орхиэктомии (удаления) — можно провести имплантацию искусственного тестикула. В некоторых случаях может быть выполнена аллопластическая пересадка донорского органа (трансплантация). Абсолютным показанием для хирургического вмешательства служит наличие опухоли. После операции назначается заместительная терапия (тестостерон).
После лечения односторонней формы заболевания пациент может вернуться к сексуальным отношениям, появляется возможность зачатия ребенка. При выраженном двустороннем поражении диагностируется бесплодие.
Лечение
В основном для ликвидации проблемы недоразвитости яичек используются методы, которые включают прием медикаментов гормонального характера, стимуляционной терапии гормонами. В зависимости от того, односторонняя либо же двусторонняя гипоплазия диагностирована у пациента, назначается специфическое лечение, которое сопровождается постоянным мониторингом состояния здоровья больного.
Если же осмотр и диагностика показывают, что одно из яичек полностью переняло на себя функции недоразвитого, гиперплазию лечат хирургическим удалением этого яичка. Процедура называется орхиэктомия, после нее, если этого захочет пациент, могут провести эстетическую хирургическую операцию, протезирование или имплантацию искусственного яичка.
В том случае, если существует дисбаланс в гормональном фоне пациента при гипоплазии, назначают трансплантацию донорского яичка или аллопластическую пересадку.
Профилактика
Профилактика гипоплазии яичка состоит в исключении отрицательного воздействия различных негативных факторов на организм беременной женщины. Кроме того, особое внимание следует уделять лечению других заболеваний, которые могут вызвать гипоплазию яичка (болезни эндокринной системы, опухоли надпочечников и др.). Для предотвращения заболевания наследственного или генетического характера семейной паре рекомендуется пройти консультацию генетика. Также с целью своевременной диагностики возможных нарушений необходимы профилактические осмотры детей и подростков такими специалистами, как уролог, эндокринолог, хирург.
Виды оказываемых услуг
| Наименование услуги | Стоимость, руб. |
| Спермограмма | 1 990 |
| MAR тест | 1 000 |
| Биопсия яичка | 20 000 |
| Неспецифическая стимуляция сперматогенеза (1 процедура) — 1-й комплекс | 1 500 |
| Неспецифическая стимуляция сперматогенеза (1 процедура) — 2-ой комплекс | 1 900 |
| УЗИ урологическое экспертное | 2 500 |
Подпишитесь на новости
Упростите свою жизнь – наша рассылка станет полезной для Вас.