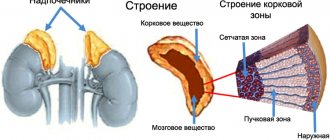
Аутоиммунная гемолитическая анемия
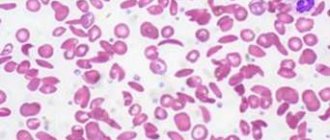
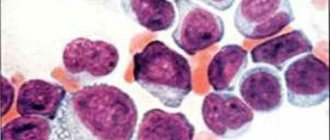
При остром начале аутоиммунных гемолитических анемий у больных появляются быстро нарастающая слабость, одышка и сердцебиение, боли в области сердца, иногда в пояснице, повышение температуры тела и рвота, интенсивная желтуха. При хроническом течении процесса отмечают относительно удовлетворительное самочувствие больных даже при глубокой анемии, нередко выраженную желтуху, в большинстве случаев увеличение селезенки, иногда и печени, чередование периодов обострения и ремиссии. Анемия носит нормохромный, иногда гиперхромный характер, при гемолитических кризах обычно отмечается выраженный или умеренный ретикулоцитоз. В периферической крови обнаруживается макроцитоз и микросфероцитоз эритроцитов, возможно появление нормобластов. СОЭ в большинстве случаев увеличена. Содержание лейкоцитов при хронической форме бывает нормальным, при острой — встречается лейкоцитоз, достигающий иногда высоких цифр со значительным сдвигом лейкоцитарной формулы влево. Количество тромбоцитов обычно нормальное. При синдроме Фишера — Ивенса аутоиммунная гемолитическая анемия сочетается с аутоиммунной тромбоцитопенией. В костном мозге эритропозз усилен, редко выявляются мегалобласты. У большинства больных снижена осмотическая резистентность эритроцитов, что обусловлено значительным числом микросфероцитов в периферической крови. Содержание билирубина увеличено за счет свободной фракции, повышено и содержание стеркобилина в кале.
Неполные тепловые агглютинины обнаруживаются с помощью прямой пробы Кумбса с поливалентной антиглобулиновой сывороткой. При положительном тесте с помощью антисывороток к IgG, IgM и т. д. уточняется, к какому классу иммуноглобулинов относятся выявляемые антитела. Если на поверхности эритроцитов менее 500 фиксированных молекул IgG, проба Кумбса отрицательна. Подобное явление наблюдается обычно у больных с хронической формой аутоиммунной гемолитической анемии или перенесших острый гемолиз. Кумбс-негативными оказываются и случаи, когда на эритроцитах фиксированы антитела, принадлежащие к IgA или IgM (в отношении которых поливалентная антиглобулиновая сыворотка менее активна). Примерно в 50% случаев идиопатических аутоиммунных гемолитических анемий одновременно с появлением иммуноглобулинов, фиксированных на поверхности эритроцитов, выявляются антитела к собственным лимфоцитам.
Гемолитическая анемия, обусловленная тепловыми гемолизинами, встречается редко. Для нее характерны гемоглобинурия с выделением мочи черного цвета» чередование периодов острого гемолитического криза и ремиссий. Гемолитический криз сопровождается развитием анемии, ретикулоцитоза (в отдельных случаях тромбоцитоза) и увеличением селезенки. Отмечаются повышение уровня свободной фракции билирубина, гемосидеринурия. При обработке донорских эритроцитов папаином удается обнаружить у больных монофазные гемолизины. У некоторых пациентов оказывается положительной проба Кумбса.
Гемолитическая анемия, обусловленная Холодовыми агглютининами (холодовая гемагглютининовая болезнь) имеет хроническое течение. Она развивается при резком повышении титра Холодовых гемагглютининов. Различают идиопатические и симптоматические формы заболевания. Ведущим симптомом болезни является чрезмерно повышенная чувствительность к холоду, которая проявляется в виде посинения и побеления пальцев рук и ног, ушей, кончика носа. Расстройства периферического кровообращения приводят к развитию синдрома Рейно, тромбофлебитов, тромбозов и трофических изменений вплоть до акрогангрены, иногда холодовой крапивницы. Возникновение вазомоторных нарушений связано с образованием при охлаждении крупных внутрисосудистых конгломератов из агглютинированных эритроцитов с последующим спазмом сосудистой стенки. Эти изменения сочетаются с усиленным преимущественно внутриклеточным гемолизом. У части больных встречается увеличение печени и селезенки. Наблюдаются умеренно выраженная нормохромная или гиперхромная анемия, ретикулоцитоз, нормальное количество лейкоцитов и тромбоцитов, увеличение СОЭ, незначительное повышение уровня свободной фракции билирубина, высокий титр полных Холодовых агглютининов (выявляемый методом агглютинации в солевой среде), иногда признаки гемоглобинурии. Характерной является агглютинация эритроцитов in vitro, возникающая при комнатной температуре и исчезающая при подогревании. При невозможности выполнения иммунологических тестов диагностическое значение приобретает провокационная проба с охлаждением (в сыворотке крови, полученной от перетянутого жгутом пальца после опускания его в ледяную воду, определяется повышенное содержание свободного гемоглобина).
При холодовой гемагглютининовой болезни в отличие от пароксизмальной холодовой гемоглобинурии гемолитический криз и вазомоторные нарушения возникают только от переохлаждения тела и гемоглобинурия, начавшаяся в условиях холода, прекращается с переходом больного в теплое помещение.
Симптомокомплекс, свойственный холодовой гемагтлютининовой болезни, может возникнуть на фоне различных острых инфекций и некоторых форм гемобластозов. При идиопатических формах заболевания полного выздоровления не наблюдается, при симптоматических прогноз зависит главным образом от тяжести основного процесса.
Пароксизмальная холодовая гемоглобинурия относится к числу редких форм гемолитических анемий. Ею заболевают люди обоего пола, чаще дети.
У больных с пароксизмальной холодовой гемоглобинурией после пребывания на холоде могут появиться общее недомогание, головная боль, ломота в теле и другие неприятные ощущения. Вслед за этим начинается озноб, повышается температура, отмечается тошнота и рвота. Моча приобретает черную окраску. Одновременно иногда выявляются желтушность, увеличение селезенки и вазомоторные нарушения. На фоне гемолитического криза у больных обнаруживают умеренную анемию, ретикулоцитоз, повышение содержания свободной фракции билирубина, гемосидеринурию и протеинурию.
Окончательный диагноз пароксизмальной холодовой гемоглобинурии устанавливают на основании обнаруженных двухфазных гемолизинов по методу Доната — Ландштейнера. Для нее не характерна аутоагглютинация эритроцитов, постоянно наблюдающаяся при холодовой гемагтлютинацией ной болезни.
Гемолитическая анемия, обусловленная эритроопсонинами. Существование аутоопсонинов к клеткам крови является общепризнанным. При приобретенной идиопатической гемолитической анемии, циррозе печени, гипопластической анемии с гемолитическим компонентом и лейкозах обнаружен феномен аутоэритрофагоцитоза.
Приобретенная идиопатическая гемолитическая анемия, сопровождающаяся положительным феноменом аутоэритрофагоцитоза, имеет хроническое течение. Периоды ремиссии, длящиеся иногда значительное время, сменяются гемолитическим кризом, характеризующимся иктеричностью видимых слизистых оболочек, потемнением мочи, анемией, ретикулоцитозом и повышением непрямой фракции билирубина, иногда увеличением селезенки и печени.
При идиопатических и симптоматических гемолитических анемиях выявление аутоэритрофагоцитоза при отсутствии данных, указывающих на наличие других форм аутоиммунных гемолитических анемий, дает основание отнести их к гемолитической анемии, обусловленной эритроопсонинами. Диагностическая проба аутоэритрофагоцитоза проводится в прямом и непрямом вариантах.
Иммуногемолитические анемии, обусловленные применением лекарств. Различные лечебные препараты (хинин, допегит, сульфаниламиды, тетрациклин, цепорин и др.), способные вызывать гемолиз, образуют комплексы со специфическими гетероантителами, затем оседают на эритроциты и присоединяют к себе комплемент, что приводит к нарушению мембраны эритроцитов. Такой механизм медикаментозно обусловленных гемолитических анемий подтверждается обнаружением на эритроцитах больных комплемента при отсутствии на них иммуноглобулинов. Анемии характеризуются острым началом с признаками внутрисосудистого гемолиза (гемоглобинурия, ретикулоцитоз, повышение содержания свободной фракции билирубина, усиление эритропоэза). На фоне гемолитического криза иногда развивается острая почечная недостаточность.
Несколько по-иному протекают гемолитические анемии, развивающиеся при назначении пенициллина и метилдофа. Введение за сутки 15 000 и более ЕД пенициллина может привести к развитию гемолитической анемии, характеризующейся внутриклеточным гипергемолизом. Наряду с общими клинико-лабораторными признаками гемолитического синдрома обнаруживается также положительная прямая проба Кумбса (выявляемые антитела относятся к IgG). Пенициллин, связываясь с антигеном мембраны эритроцитов, образует комплекс, против которого в организме вырабатываются антитела.
При длительном применении метилдофа у части больных возникает гемолитический синдром, имеющий черты идиопатической формы аутоиммунных гемолитических анемий. Выявляемые антитела идентичны с тепловыми агглютининами и относятся к IgG.
Гемолитическая анемия, обусловленная механическими факторами, связана с разрушением эритроцитов при их прохождении через измененные сосуды или через искусственные клапаны. Эндотелий сосудов изменяется при васкулитах, злокачественной артериальной гипертензии; при этом адгезия и агрегация тромбоцитов активированы, как и система свертывания крови и образования тромбина. Развиваются распространенный стаз крови и тромбоз мелких кровеносных сосудов (ДВС-синдром) с травматизацией эритроцитов, в результате чего они фрагментируются; в мазке крови находят многочисленные фрагменты эритроцитов (шистоциты). Разрушаются эритроциты также при их прохождении через искусственные клапаны (чаще — при многоклапанной коррекции); описана гемолитическая анемия на фоне сенильного кальцинированного аортального клапана. Диагноз базируется на признаках анемии, повышении концентрации свободного билирубина в сыворотке крови, наличии шистоцитов в мазке периферической крови и симптоматике основного заболевания, ставшего причиной механического гемолиза.
Реже встречается в клинической практике гемолитическая анемия, обусловленная воздействием свинца, при отравлении кислотами, ядами змей или дефицитом витамина Е, а также внутриклеточными паразитами. Гемолитическая анемия развивается, например, после укуса змеи, случайного или намеренного (суицид) приема уксусной кислоты, при контакте с парами свинца, на фоне малярии. Анемия носит нормоцитарный, нормохромный, регенераторный характер; в сыворотке крови повышено содержание свободной фракции билирубина и железа.
Гемолитико-уремический синдром (болезнь Мошковича, синдром Гассера) может осложнять течение аутоиммунных гемолитических анемий. Заболевание аутоиммунной природы характеризуется гемолитической анемией, тромбоцитопенией, поражением почек. Отмечаются диссеминированное поражение сосудов и капилляров с вовлечением практически всех органов и систем, выраженные изменения со стороны коагулограммы, характерные для ДВС-синдрома.
Публикации в СМИ
Гемолитические анемии — большая группа анемий, характеризующихся снижением средней продолжительности жизни эритроцитов (в норме 120 дней). Гемолиз (разрушение эритроцита) может быть внесосудистым (в селезёнке, печени или костном мозге) и внутрисосудистым. Общие признаки — выраженная интоксикация с ознобом и лихорадкой, боли в пояснице и животе, возможен шок в результате нарушения микроциркуляции, желтуха, спленомегалия, гемоглобинурия.
Этиология. Гемолитические анемии возникают при дефектах эритроцитов (внутриклеточные факторы) либо под воздействием внешних по отношению к эритроцитам причин (внеклеточные факторы). Обычно внутриклеточные факторы — наследуемые, а внеклеточные — приобретённые.
• Внеклеточные факторы. Микроокружение эритроцитов представлено плазмой и эндотелием сосудов. Присутствие в плазме токсических веществ или инфекционных агентов вызывает изменения стенки эритроцита, что приводит к: (1) выработке аутоантител к изменённому эритроциту (классический пример — аутоиммунная гемолитическая анемия), (2) непосредственному разрушению эритроцита •• Изоиммунные гемолитические анемии наблюдают при эритробластозе плода; сюда также можно отнести гемолитические трансфузионные реакции •• Дефекты эндотелия сосудов (микроангиопатии) также способны повреждать эритроциты — анемия гемолитическая микроангиопатическая. У детей может протекать остро в виде гемолитико-уремического синдрома •• Гемоглобинурия пароксизмальная холодовая •• Назначение некоторых ЛС (например, сульфаниламидов, противомалярийных препаратов) приводит к гемолитическому кризу.
• Внутриклеточные факторы. Внутриклеточные дефекты включают аномалии мембран эритроцитов, Hb или ферментов. Эти дефекты наследуемы (исключая пароксизмальную ночную гемоглобинурию) •• Дефекты мембран ••• Сфероцитоз наследуемый ••• Эллиптоцитоз наследуемый ••• Гемоглобинурия пароксизмальная ночная •• Гемоглобинопатии (например, серповидноклеточная анемия). Известно более 300 заболеваний, обусловленных точечными мутациями генов глобинов. Дефект молекулы глобина способствует нарушению его полимеризации. Изменяются мембрана, форма эритроцита, увеличивается подверженность гемолизу •• Энзимопатии.
Некоторые гемолитические анемии, обусловленные недостаточностью ферментов
• Анемия вследствие недостаточности глюкозо-6-фосфатдегидрогеназы (Г-6-ФД, см. также приложение к этой статье). Клиническая картина: острые эпизоды гемолитической анемии, обычно провоцируемые ЛС, инфекциями (также при желтухе новорождённых и фавизме). Лечение • Заместительная терапия (гемотрансфузии) • Спленэктомия при недостаточности Г-6-ФД бесполезна • Пациенты с вариантами недостаточности Г-6-ФД, связанными с острыми гемолитическими кризами, должны избегать приёма ЛС, вызывающих гемолиз. МКБ-10 D55.0 Анемия вследствие недостаточности Г-6-ФД.
• Анемия вследствие недостаточности пируват киназы. Недостаточность пируват киназы — вторая по частоте (после недостаточности Г-6-ФД) наследуемая энзимопатия эритроцитов. Пируват киназа катализирует конечный этап гликолиза; последствие её недостаточности — неадекватная продукция аденозинтрифосфорной кислоты (АТФ), что негативно отражается на функциях эритроцитов, в т.ч. на работе Na+-K+-АТФазы — дефектные клетки теряют К+. Особенно интенсивному разрушению подвергаются ретикулоциты (преимущественно в селезёнке). Клиническая картина • Возможны проявления гемолиза различной степени тяжести; у новорождённых — желтуха, хронический гемолиз, спленомегалия • Симптомы недостаточности пируват киназы идентичны таковым при прочих хронических гемолитических анемиях • Из-за избирательного разрушения ретикулоцитов их количество в периферической крови может не отражать тяжести анемии. Лечение: эффективна спленэктомия. МКБ-10 D55.2 Анемия вследствие нарушений гликолитических ферментов.
• Гемолитическая анемия развивается также при болезни Норума, а также вследствие недостаточности эритроцитарной формы аденилат киназы (*103000, КФ 2.7.4.3, ген АК1, 9q34.1, r), аденозинтрифосфатазы (*102800, АТФаза, КФ 3.6.1.3, r, также Â), d-аминолевулинат дегидратазы (*125270, порфобилиноген синтетаза, КФ 4.2.1.24, 9q34, ген ALAD, не менее 7 мутантных аллелей, наследуемых как r, так и Â), гексокиназы 1 (КФ 2.7.1.1, *142600, локус 10q22, ген HK1, r), g-глутамилцистеин синтетазы (*230450, КФ 6.3.2.2, r], глутатион пероксидазы 1 (КФ 1.11.1.9, *138320, GPX1, 3q11–q12, r), глутатион редуктазы (*138300, КФ 1.6.4.2, 8p21.1, ген GSR, r), глюкозо-6-фосфат изомеразы (*172400, КФ 5.3.1.9, 19q13.1, ген GPI, Â, r), 2,3-дифосфоглицерат мутазы (222800, КФ 5.4.2.4, 7q31–q34, ген BPGM, r), енолазы (*172430, КФ 4.2.1.11, ген ENO1), копропорфириноген оксидазы (*121300, КФ 1.3.3.3, 3q12, ген CPO, Â), пиримидин нуклеотидазы (*266120, КФ 3.1.3.5, ген P5N, r), триозофосфат изомеразы (*190450, КФ 5.3.1.1, 12p13, ген TPI1), феррохелатазы (*177000, КФ 4.99.1.1, 18q21.3, ген FECH, Â, r), фосфоглицерат киназы (*311800, КФ 2.7.2.3, À [Xq13, ген PGKА]; также Â [хр. 19, ген PGKВ]), 6-фосфоглюконолактоназы (*172150, КФ 3.1.1.31), фосфофруктокиназы (*171860, КФ 2.7.1.11, 21q22.3, ген PFKL). Лечение индивидуальное в зависимости от вида гемолитической анемии (например, спленэктомия или заместительная терапия препаратами железа).
МКБ-10 • D55 Анемия вследствие ферментных нарушений • D56 Талассемия • D57 Серповидноклеточные нарушения • D58 Другие наследственные гемолитические анемии • D59 Приобретённая гемолитическая анемия
ПРИЛОЖЕНИЯ
Глюкозо-6-фосфат дегидрогеназа (Г-6-ФД, КФ 1.1.1.49, *305900, Xq28) важна для поддержания внутриклеточного содержания восстановленных нуклеотидов; наследуемая недостаточность фермента (À, крайний полиморфизм, более трёхсот аллельных вариантов) приводит к развитию анемий, фавизму, хронической гранулематозной болезни. Различают варианты фермента: вариант А и средиземноморский • Вариант А встречают у афроамериканцев (быстро деградирующий изофермент, время полужизни — 13 дней) • Средиземноморский вариант обнаруживают в основном у греков и итальянцев (крайняя нестабильность фермента, время полужизни — несколько часов), фактически активность изофермента отсутствует • При варианте А или средиземноморском варианте недостаточности Г-6-ФД приём ЛС, проявляющих окислительные свойства (сульфаниламиды, салицилаты, фенацетин), через 1–3 дня инкубационного периода вызывает острейший гемолитический криз. Последующее течение заболевания различно для двух вариантов фермента •• Вариант А. Происходит гемолиз предсуществовавшей эритроцитарной популяции. Гемолитический криз разрешается при образовании в костном мозге новых популяций эритроцитов с высокой активностью Г-6-ФД •• Средиземноморский вариант. Гемолиз приводит к разрушению почти всех эритроцитов. Показана трансфузия вплоть до полного выведения ЛС из организма.
Фавизм (примахиновая анемия) — острое состояние, возникающее при употреблении в пищу некоторых видов бобовых (например, конских бобов Vicia fava), в результате ингаляции пыльцы их цветов, а также после приёма некоторых ЛС (примахин, сульфаниламиды, салицилаты, нитрофураны, производные витамина К и др.). Встречается повсеместно, но чаще в районах, эндемичных по малярии. Возможен у части лиц с наследственной недостаточностью Г-6-ФД (305900, ген G6PD, Xq28). Клиническая картина: высокая температура тела, головная боль, боли в животе, гемолитическая анемия, эозинофилия, желтуха, диарея, рвота, упадок сил и кома. Лечение поддерживающее • гемотрансфузии • фолиевая кислота • поддержание адекватного диуреза • алкалинизация мочи. МКБ-10. D55.0 Анемия вследствие недостаточности глюкозо-6-фосфатдегидрогеназы. Фавизм.
Гемоглобинурия пароксизмальная ночная — хроническое заболевание с эпизодами гемолитической анемии и гемоглобинурией (преимущественно по ночам). Характерны желтушность кожи или её бронзовая окраска, умеренное увеличение селезёнки, иногда — печени, макроцитоз, анизоцитоз. Наблюдают редко (2/1 млн населения). Предрасположенность к развитию заболеванию (*311770, Xq22.1, ген PIGA, À). Диагностика: эпизоды утренней чёрной мочи, при добавлении столового сахара к крови больного происходит гемолиз. Лечение. ГК (например, преднизолон 20–40 мг/сут) вызывают стойкую ремиссию у 50% пациентов. Не рекомендуют переливать плазму, но отмытую эритроцитарную массу можно переливать во время кризов. Гепарин следует применять с осторожностью, т.к. возможно усиление гемолиза. Препараты железа внутрь. Синонимы: синдром Маркиафавы–Микели, болезнь Маркиафавы–Микели. МКБ-10. D59.5 Пароксизмальная ночная гемоглобинурия. Холодовая — редкое заболевание, характеризующееся возникновением гемолиза после холодового воздействия (атмосферный воздух, холодная вода). Обычно возникает после перенесённой вирусной инфекции. Гемолиз индуцируется фиксированными на эритроцитах гемолизинами Доната–Ландштайнера. Течение может быть острым, прогрессирующим или рецидивирующим. Синонимы: анемия гемолитическая Доната–Ландштайнера, синдром Дресслера, синдром Гарли. МКБ-10. D59.6 Гемоглобинурия вследствие гемолиза, вызванного другими внешними причинами.
Стоматоцитоз • I (#185020, 9q34.1, ген EPB72 [133090], Â), мутация гена стоматина (внутренний белок мембран эритроцитов). Клинически: гемолитическая анемия, стоматоцитоз. Лабораторно: укорочение времени циркуляции эритроцитов, их увеличенная осмотическая резистентность, увеличение содержания внутриклеточного содержания ионов натрия • Стоматоцитоз II (*185010, Â). Клинически: гемолитическая анемия, стоматоцитоз, холелитиаз, периодическая желтуха. Лабораторно: уменьшенная осмотическая резистентность эритроцитов, увеличение внутриклеточного натрия • Стоматоцитоз холодочувствительный (185020, Â). Клинически: гемолитическая анемия, стоматоцитоз. Лабораторно: Усиление аутогемолиза и увеличение осмотической резистентности эритроцитов при 5 °C, холодовой гемолиз, предотвращаемый уменьшением pH или увеличением содержания АТФ.
Недостаточность альдолазы. Фруктозо-1,6-дифосфат альдолаза (триозофосфатлиаза, КФ 4.1.2.13, 3 изофермента: альдолазы 1, 2 и 3, или А, B и C) — гликолитический фермент, катализирующий обратимое превращение фруктозо-1,6-дифосфата в глицеральдегид 3-фосфат и дигидроксиацетон фосфат. Альдолаза A экспрессируется в тканях плода и в скелетных мышцах (5% от всего мышечного белка), альдолаза В — в печени, почках, кишечнике, альдолазы A и C — в нервной ткани. Известны наследственные заболевания, развивающиеся вследствие недостаточности разных альдолаз. Альдолаза А (103850, 16q22–q24, ген ALDOА, r). При недостаточности фермента развивается врождённая несфероцитарная гемолитическая анемия (желтуха, спленомегалия, холелитиаз, возможны низкорослость, задержка умственного развития и полового созревания). Альдолаза В (229600, 9q22, ген ALDOВ, r). При мутациях гена развивается врождённая непереносимость фруктозы (фруктоземия). Клинически: вялость сосания, задержка роста и развития, гипогликемия, метаболический ацидоз, рвота, гепатомегалия, цирроз печени, желудочно-кишечные кровотечения, судорожные припадки, проксимальный канальцевый ацидоз. Лабораторно: фруктоземия, гипербилирубинемия, гиперурикемия, глюкозурия, гипофосфатемия и фосфатурия, высокие значения pH мочи. МКБ-10. D55.2 Анемия вследствие нарушений гликолитических ферментов.