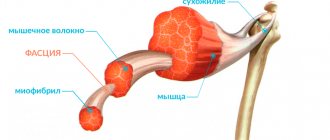
Мышечная система выполняет ряд важнейших функций в организме человека, от ее правильного функционирования напрямую зависят двигательные способности, координация движений. Патологические процессы, сопровождающиеся поражением мышечных структур, крайне тяжело переносятся человеком и в большинстве случаев приводят к инвалидизации. Одной из наиболее тяжелых патологий такого характера является миопатия Дюшенна.
В клиниках «Вивантес» возможна только диагностика этого заболевания.
В Германии диагностика и лечение миопатии Дюшенна проводится квалифицированными врачами высшей категории. Специалисты проводят полный спектр исследований для верификации заболевания. На основе результатов диагностики индивидуально разрабатывается тактика лечения, которая базируется на инновационных методиках консервативной терапии.
Что такое миопатия Дюшенна
Миопатия или прогрессирующая мышечная дистрофия Дюшенна – патологический процесс, характеризующийся поражением мышечных структур организма и приводящий к слабости мышц. Одной из основных особенностей заболевания является факт постоянного, постепенного прогрессирования. Болезнь дебютирует и обнаруживается в первые годы жизни человека, преимущественно у детей в возрасте от 2 до 5 лет.
Характерной особенностью миопатии Дюшенна выступает тот факт, что это заболевание передается генетически. Наследственная патология скреплена с Х-хромосомой, то есть она передается ребенку от матери. При этом женщины, от которых дети наследуют заболевание, никогда им не болеют.
Патологический процесс сопровождается дегенеративным поражением мышечных структур. Миопатия Дюшенна характерна не только ранним возникновением, болезнь склонна к стремительному прогрессированию. В результате у ребенка развивается мышечная слабость, последствиями которой становятся различные скелетные деформации. На поздних стадиях прогрессирования имеет место поражение сердечной мышцы и сопутствующая сердечная недостаточность.
Истинная причина развития мышечной дистрофии Дюшенна заключается в наследственной генетической мутации. При этом мутации делятся на две группы:
- В 70% случаев причина заключается в дефекте гена дистрофина, унаследованного от матери;
- В остальных 30% случаев развитие болезни Дюшенна обусловлено локальными мутациями яйцеклеток матери.
Тяжелое течение патологического процесса обусловлено преимущественно полным прекращением синтеза дистрофина. В результате мышцы утрачивают сократительную способность, подвержены снижению тонуса и общей силы.
Литература
- van Putten M., Hulsker M., Nadarajah V.D., van Heiningen S.H., van Huizen E., van Iterson M. et al. (2012). The effects of low levels of dystrophin on mouse muscle function and pathology. PLoS One. 7, e31937;
- Russo F.B., Cugola F.R., Fernandes I.R., Pignatari G.C., Beltrão-Braga P.C. (2015). Induced pluripotent stem cells for modeling neurological disorders. World J. Transplant. 5, 209–221;
- Falzarano M.S., Scotton C., Passarelli C., Ferlini A. (2015). Duchenne muscular dystrophy: from diagnosis to therapy. Molecules. 20, 18168–18184;
- Bushby K., Finkel R., Birnkrant D.J., Case L.E., Clemens P.R., Cripe L. et al. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9, 77–93;
- Bushby K., Finkel R., Birnkrant D.J., Case L.E., Clemens P.R., Cripe L. et al. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 9, 177–189;
- Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E. et al. (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 351, 400–403;
- Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M. et al. (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 351, 403–407;
- Tabebordbar M., Zhu K., Cheng J.K., Chew W.L., Widrick J.J., Yan W.X. et al. (2016). In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 351, 407–411;
- Элементы: «Прокариотическая система иммунитета поможет редактировать геном»;
- Gallagher J. (2016). Scientists get ’gene editing’ go-ahead. BBC News;
- CRISPR-системы: иммунизация прокариот;
- Мутагенная цепная реакция: редактирование геномов на грани фантастики;
- А не замахнуться ли нам на… изменение генома?;
- Нобелевская премия по физиологии и медицине (2012): индуцированные стволовые клетки;
- Болезнь Альцгеймера: ген, от которого я без ума;
- Как спасти Тринадцатую? (Перспективы лечения болезни Хантингтона);
- McGreevy J.W., Hakim C.H., McIntosh M.A., Duan D. (2015). Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis. Model. Mech. 8, 195–213;
- Singh S.M., Kongari N., Cabello-Villegas J., Mallela K.M. (2010). Missense mutations in dystrophin that trigger muscular dystrophy decrease protein stability and lead to cross-β aggregates. Proc. Natl. Acad. Sci. USA. 107, 15069–15074;
- Goyenvalle A., Seto J.T., Davies K.E., Chamberlain J. (2011). Therapeutic approaches to muscular dystrophy. Hum. Mol. Genet. 20, R69–R78.
Симптомы и диагностика
Клиническая картина миопатии Дюшенна становится все более разнообразной по мере прогрессирования заболевания. При этом мышечная слабость начинается с нижних конечностей, постепенно распространяясь вверх на бедра, тазовые мышцы. В силу того, что дебютирует патология в раннем возрасте, первые признаки выражаются в конкретных отставаниях ребенка в развитии. К числу основных симптомов болезни Дюшенна специалисты относят:
- нарушения походки у детей, при этом ребенок поздно начинает ползать и ходить;
- в попытках встать, ребенок явно опирается на руки или встает с колена;
- формирование «осиной» талии вследствие мышечной атрофии;
- постепенное выпадение сухожильных рефлексов;
- присутствуют различные формы искривления позвоночного столба (кифоз, лордоз);
- деформация грудной клетки (например, килевидное строение);
- расстройства со стороны сердечно-сосудистой системы;
- как правило, к 12 годам больной утрачивает способность ходить, а к 15 самостоятельно выполнять большинство других движений;
- также в клинической картине присутствуют различные формы умственной отсталости, включая аутизм и другие расстройства.
Приступая к диагностике, врачи придают особое значение фиксации всех имеющихся симптомов, изучению анамнеза, общему осмотру пациента. Помимо этого, проводятся лабораторные и аппаратные исследования, в числе которых наиболее актуальны:
- развернутое исследование крови;
- общеклиническое исследование мочи;
- биохимическое исследование крови;
- рентгенологические исследования;
- УЗИ органов брюшной полости, забрюшинного пространства, сердца;
- эхокардиография;
- исследование на определение концентрации дистрофина в мышечных тканях;
- биопсия мышц;
- консультации профильных специалистов (кардиолог, генетик).
«Амондис 45»: безопасность касимерсена в лечении мышечной дистрофии Дюшенна
Исследования на самцах мышей и крыс засвидетельствовали почечную токсичность касимерсена, проявившуюся дегенерацией почечных канальцев. И хотя испытания на людях не отметились ничем таким подобным, необходимо принимать во внимание, что в ходе изучения некоторых других антисмысловых олигонуклеотидов пациенты сталкивались с почечной токсичностью, включая потенциально летальный гломерулонефрит. Потому терапия «Амондисом 45» должна сопровождаться мониторингом функции почек.
Среди наиболее частых побочных реакций на назначение касимерсена: инфекции верхних дыхательных путей (у 65% пациентов — против 55% в группе плацебо), кашель (33% против 26%), лихорадочное состояние (33% против 23%), головная боль (32% против 19%), суставная боль (21% против 10%), боль в ротоглотке (21% против 7%).
Лечение миопатии Дюшенна
Основные принципы лечения миопатии Дюшенна основаны на применении консервативной терапии, медикаментозной и физиотерапевтических процедур. План лечения для каждого пациента разрабатывается индивидуально. При этом специалисты учитывают присутствующие симптомы, результаты диагностики, возраст пациента, степень прогрессирования мышечной дистрофии.
Главная цель врачей при лечении миопатии Дюшенна – максимальное замедление прогрессирования дистрофических процессов и улучшение качества жизни пациента. В рамках комплексной медикаментозной терапии основными группами препаратов являются:
- Глюкокортикостероиды – действие этих средств направлено на замедление развития болезни, на ранних стадиях применяемые препараты последнего поколения могут даже остановить развитие патологии на некоторое время.
- Прозерин – специализированный препарат, способствующий улучшению передачи нервных импульсов в мышечных структурах, благодаря чему удается поддерживать тонус мышц.
- Препараты кальция – обеспечивают поддержание нормального сокращения мышечных волокон.
- Витаминные комплексы – применяются преимущественно для улучшения обменных процессов, что также способствует поддержанию мышечного тонуса.
Что касается физиотерапии, специалисты в Германии индивидуально разрабатывают программы ЛФК и массажа, назначают ношение специальных поддерживающих шин. Учитывая уникальность каждого клинического случая, специалисты предпринимают все возможные меры, чтобы помочь пациенту, повысить качество и максимально увеличить продолжительность жизни, ограничив прогрессирование болезни.
В клиниках «Вивантес» возможна только диагностика этого заболевания.
Результаты
У 63% больных с МДД заболевание дебютировало в возрасте 2—5 лет, у 37% — в 5—7 лет. У всех больных заболевание начиналось с поражения мышц тазового пояса и проксимальных отделов ног и носило неуклонно прогрессирующий характер. Основными клиническими симптомами были слабость и атрофия мышц ног и тазового пояса с развитием псевдогипертрофий икроножных мышц (рис. 1).
Рис. 1. Больной С., 5 лет. Диагноз МДД, установлена делеция в 52-м экзоне, псевдогипертрофия икроножных мышц. Выявлялись контрактуры в коленных и голеностопных суставах, у части больных отмечались выраженные деформации позвоночника (рис. 2).
Рис. 2. Больной Р., 15 лет. Диагноз МДД, установлена делеция в 44—50-м экзоне, выраженный кифосколиоз. Клинические симптомы кардиопатии наблюдались у 68% больных, пневмопатии — у 53%, 13% пациентов на момент обследования находились на искусственной вентиляции легких. Нейропсихологическое исследование выявило в 33% случаев выраженные когнитивные расстройства, в 19% — умеренные нарушения когнитивной сферы.
Данные иммуноферментного анализа нейротрофинов свидетельствуют, что концентрации ФРН в сыворотке крови у больных с МДД выше, чем в контрольной группе, — 2391 [1587; 4136] и 553 пг/мл [314; 864] соответственно (р
<0,001).
Молекулярно-генетическое исследование установило в 58% случаев наличие делеций и дупликаций, в 8% — нонсенс-мутаций в различных экзонах. Распределение мутаций в гене МДД носило неравномерный характер, наиболее часто они встречались на участке гена с 43-й по 50-й экзон (рис. 3).
Рис. 3. Распределение больных по мутациям в гене МДД по экзонам. По оси абсцисс — экзоны, по оси ординат — число больных.
Анализ места расположения мутации в гене МДД позволил выявить, что имеет место нарушенный синтез изоформ белка дистрофина Dp260 (n
=17), изоформ Dp140 (
n
=15), Dp116 (
n
=1) и Dp71 (
n
=1). У 15 больных отмечалась мутация в гене МДД в экзонах, влияющих на синтез двух (Dp260 и Dp140) (
n
=14) и более изоформ (Dp140, Dp260, Dp116 и Dp71) (
n
=1) (табл. 1).
Таблица 1. Основные молекулярно-генетические и клинические характеристики обследованных больных МДД Примечание. Обозначение 5’UTR означает, что у гена Dp140 повреждается 5’-нетранслируемая область, которая белок не кодирует, но содержит регуляторные элементы. Вследствие того, что 5’UTR очень протяженная (около 1000 kb), не все мутации внутри нее могут иметь одинаковое повреждающее значение.
Полученные результаты клинико-неврологического, молекулярно-генетического и лабораторного исследований были сопоставлены с целью уточнения роли генетических и биохимических факторов в развитии когнитивных расстройств при МДД. Было выявлено достоверное снижение концентрации нейротрофина ФРГМ во 2-й группе. Проверка эмпирического распределения переменных, характеризующих концентрацию нейротрофинов, в соответствии с законом нормального распределения по критерию Шапиро—Уилка показала достоверное различие (p
<0,01). Таким образом, для описания числовых характеристик указанных количественных признаков наиболее адекватно использование медианы и квартилей: Me [Q25%; Q75%], здесь и далее средняя тенденция количественной случайной величины будет описываться медианой, а особенности закона распределения уточняться границами интерквартильного размаха (рис. 4).
Рис. 4. Концентрация ФРГМ, пг/мл (ось ординат), в сыворотке крови в исследуемых группах (ось абсцисс).
Таким образом, концентрация ФРГМ имеет статистические значимые различия в группах пациентов с наличием и отсутствием когнитивных расстройств (p
<0,001). Во 2-й группе наблюдалась сниженная концентрация ФРГМ — 23 670 [21 700; 30 720] пг/мл против 32 700 [31 660; 33 750] пг/мл в 1-й группе. Для данного показателя был выполнен расчет абсолютного риска варианта течения МДД с когнитивными нарушениями, отношения шансов этого риска (ОШ) и 95% доверительного интервала (ДИ) О.Ш. Уровень ФРГМ был разделен на два интервала в соответствии с законом распределения этой переменной в группах. В табл. 2 представлены
Таблица 2. Оценка абсолютного риска развития когнитивных расстройств при МДД в зависимости от концентрации ФРГМ Примечание. * — группа сравнения или минимального прогнозируемого риска. результаты определения абсолютного риска течения МДД с развитием когнитивных расстройств и ОШ этого риска при оценке связи концентрации ФРГМ и течения МДД. На основании анализа закона распределения было выявлено, что минимальный риск течения МДД с развитием когнитивных расстройств, составивший 20% (2 из 10 пациентов), наблюдался у больных с концентрацией ФРГМ более 31 000 пг/мл.
По сравнению с данной группой у пациентов с концентрацией ФРГМ менее 31 000 пг/мл риск неблагоприятного течения составил 75% (15 из 20 пациентов), шансы развития когнитивных расстройств статистически значимо (p
<0,001) увеличивались более чем в 10 раз (ОШ=12,0; 95% ДИ ОШ=[1,9—76,4]).
Нейротрофины ФРН и ЦНТФ не имели достоверных различий между показателями концентрации в 1-й и 2-й группах. При исследовании концентрации нейротрофинов в зависимости от места расположения мутации в гене МДД было выявлено достоверное снижение концентрации ФРН в группе с дистальным расположением мутации. Для уточнения роли расположения мутации в гене МДД и развития когнитивных расстройств у больных было проведено сопоставление данных молекулярно-генетического и клинико-нейропсихологического исследования, которое не выявило достоверных различий по наличию когнитивных расстройств и месту расположения мутации в гене МДД. Однако более выраженные нарушения в когнитивной сфере наблюдались у больных с МДД с расположением мутации в дистальном отделе гена МДД. Изучение роли отдельных изоформ белка дистрофина в развитии когнитивных расстройств не позволило выявить достоверных данных вследствие недостаточности клинических наблюдений.
Проф. д-р Беттина Шмитц
Неврология – Эпилепсия
Заведующая отделением неврологии
Cпециализация
- Эпилепсия
- Дифференциальная диагностика неэпилептических пароксизмов
- Длительный ЭЭГ — видеомониторинг
- Медицинская и немедицинская помощь при пароксизмах и осложнениях, связанных с эпилепсией
- Член Британской Медицинской Ассоциации
- Инструктор и член Европейской Академии по изучению Эпилепсии (EUREPA)
- Член Комиссии по психобиологии и Международной Лиги по борьбе с Эпилепсией (ILAE)
- Психоорганический синдром, Всемирная Федерация Обществ Биологической Психиатрии (WFSBP)
- Видеоконсультация
Показать личностный профиль врача
Обсуждение
Клинико-нейропсихологическое исследование больных с МДД в 33% случаев выявило когнитивные нарушения различной степени выраженности. В последние годы изучение патогенеза когнитивных расстройств у больных с МДД имело приоритетное направление — выявление связи между когнитивным дефицитом и местом расположения мутации в гене МДД. Было отмечено, что среди больных с МДД при делеции 52-го экзона чаще диагностировались когнитивные расстройства [21]. Однако ряд исследователей [22] объясняют это тем, что при МДД чаще выявляются мутации в экзонах 44—53, тем самым формируется более обширная когорта больных. Среди обследованных нами больных 65% имели мутации в гене МДД, расположенные в экзонах 43—53, т. е. дистальной части гена. Известно [23], что дистальное расположение мутации в гене белка МДД является предрасполагающим фактором для развития когнитивных расстройств. В нашем исследовании более выраженные нарушения когнитивной сферы были зафиксированы также у больных с дистальным расположением мутации в гене белка МДД. В настоящее время активно изучается [24] вклад различных изоформ белка МДД в развитие когнитивных нарушений. Установлено [25, 26], что для когнитивных расстройств особенно важны изоформы дистрофина Dp140 и Dp71. По нашим данным, среди выявленных мутаций преобладали затрагивающие синтез изоформы Dp140, так как известно, что промотор изоформы Dp140 располагается в интроне 44. Необходимо отметить, что среди 4 больных МДД с выраженными нарушениями в когнитивной сфере у 3 была выявлена мутация, затрагивающая изоформы Dp260 и Dp140, и у 1 поврежден синтез изоформ Dp140, Dp260, Dp116 и Dp71. Эти данные показывают, что более выраженные когнитивные расстройства присутствуют у больных с наличием мутации в гене МДД, затрагивающей синтез нескольких изоформ белка МДД, однако малочисленность клинических наблюдений не позволяет оценить достоверность полученных результатов. Впервые была предпринята попытка оценить влияние концентрации нейротрофинов в сыворотке крови больных с МДД на развитие когнитивных расстройств и сопоставить полученные данные с результатами молекулярно-генетического анализа. Выявлено, что в группе больных с МДД с наличием когнитивных расстройств имеет место достоверное снижение концентрации нейротрофина ФРГМ. Сопоставляя собственные наблюдения с данными N. Doorenweerd и соавт. [27] по изучению микроструктуры головного мозга у больных с МДД с помощью методики количественной магнитно-резонансной томографии, мы можем гипотетически объяснить роль данного фактора в развитии когнитивного дефицита. Было отмечено, что больные с МДД с наличием мутации, затрагивающей изоформу Dp140 (–), и прошедшие нейропсихологическую экспертизу с худшими результатами, имели меньший объем головного мозга, а также серого вещества по сравнению с группами контроля и больных с МДД с Dp140 (+), получивших положительную оценку при тестировании. Те же авторы, изучая церебральный кровоток у больных с МДД, выявили [28], что часть пациентов имеют сниженную церебральную перфузию. Однако эти данные не коррелировали с уменьшением объема головного мозга и серого вещества у больных с МДД. В то же время больные с нарушениями церебральной гемодинамики имели в гене МДД мутации, затрагивающие изоформу Dp140. Было высказано предположение о влиянии изоформы Dp140 на функционирование сосудистых эндотелиальных клеток и астроцитов, опосредованно влияющих на состояние церебральной перфузии. Вероятно, уменьшение объема головного мозга можно гипотетически связать с низким уровнем нейротрофина ФРГМ, так как именно этот фактор способствует росту и дифференцировке нейронов головного мозга [19]. Нами установлено, что у пациентов с концентрацией ФРГМ менее 31 000 пг/мл риск неблагоприятного течения составил 75% (15 из 20 пациентов), шансы развития когнитивных расстройств статистически значимо (p
<0,001) увеличивались более чем в 10 раз (ОШ=12,0; 95% ДИ ОШ=[1,9—76,4]). Отметим, что у больных с МДД с дистально расположенной мутацией в гене имела место более высокая концентрация нейротрофина ФРН. Согласно результатам предыдущих исследований нейротрофической регуляции у детей с тяжелыми формами наследственной и приобретенной патологии ЦНС [29, 30], ФРН является маркером патологического процесса. Необходимо учитывать и нейритингибирующий эффект высоких концентраций ФРН, выявленный в серии экспериментов с сывороткой крови больных спинальной мышечной атрофией в органотипической культуре нервной ткани [31, 32].
Таким образом, в развитии когнитивных расстройств у больных с МДД участвует ряд механизмов, сочетание которых приводит к более выраженному когнитивному дефекту. Среди них отмечаются как молекулярно-генетические факторы — дистальное расположение мутации в гене МДД, влияние различных комбинаций нарушенного синтеза изоформ белка МДД (Dp140, Dp116, Dp260 и Dp71), так и биохимические факторы, связанные с особенностями нейротрофической регуляции, проявляющиеся сниженным уровнем нейротрофина ФРГМ в сыворотке крови у больных с МДД и повышенной концентрацией ФРН.
Авторы заявляют об отсутствии конфликта интересов.
Проф. д-р Йорг Мюллер
Неврология – Паркинсон, РС
Заведующий отделением неврологии
Cпециализация
- Признанный международный эксперт в области болезни Паркинсона, дистонии и тремор
- Нейромускулярные заболевания
- Рассеянный склероз
- Лечение с использованием бутолотоксина (дистония, спастика)
- Лечение глубокой стимуляцией мозга
- Автор более 70 научных публикаций, явялется членом международных экспертных советов
Показать личностный профиль врача
«Народные» методы лечения могут усугубить заболевание
Если диагноз подтвержден, врач назначит глюкокортикостероиды — на сегодняшний день это золотой стандарт лечения для замедления развития заболевания. Также показана специальная лечебная физкультура и комплексная реабилитация в разном возрасте. В 2020 году в России зарегистрирован первый препарат для лечения миодистрофии Дюшенна — Аталурен, но пока о его эффективности известно немного.
Создатели просветительского проекта Red Balloons для родителей детей с болезнью Дюшенна предупреждают об опасности различных методов «лечения» из Интернета, за которую отчаявшиеся мамы и папы хватаются как за соломинку. К сожалению, они не только не приносят пользу, но могут даже ускорить прогрессирование заболевания.
Чего делать нельзя ни в коем случае:
- давать чрезмерную физическую нагрузку (заниматься агрессивными видами спорта, взбираться и спускаться по крутым лестницам, прыгать на батутах);
- парить ноги, мыться очень горячей водой, ходить в сауны и бани;
- без предварительной консультации врача делать массаж;
- прибегать к способам «лечения» из интернета: стволовые клетки, пиявки и обертывания не помогают.
Проф. д-р Бруно-Марсель Макерт
Неврология
Заведующий отделением неврологии
Cпециализация
- Всемирно признанный специалист в области инсульта, заболеваний периферической нервной системы и клинической электрофизиологии
- Автор свыше 40 оригинальных публикаций
- Совет директоров Берлинского сообщества по предотвращению инсульта (BSA)
- Член совета директоров Берлинского центра исследований инсульта (CSB)
Показать личностный профиль врача
Проф. д-р Йорг Виссель, MD, FRCP
Неврологическая реабилитация
Заведующий oтделением неврологической реабилитации и физиотерапии, отделением неврологии и Центром реабилитации
Cпециализация
- Неврологическая реабилитация после инсульта, полученных черепно-мозговых травм и повреждений спинного мозга
- Реабилитационная терапия при болезни Паркинсона и дистонии
- Ботулинотерапия
- Лечение спастичности
- Неврологическая реабилитация с помощью интратекальной баклофеновой терапии и глубокой мозговой стимуляции
- Автор более 90 научных публикаций, член нескольких экспертных комиссий
Показать личностный профиль врача
Это заболевание часто путают с гепатитом
Если есть сомнения по поводу здоровья мальчика, врач должен назначить анализ крови на активность креатинфосфокиназы (КФК). Это фермент, содержащийся в скелетных мышцах, маркер их распада. При мышечных дистрофиях, в том числе болезни Беккера, он выделяется в громадных количествах — в несколько десятков тысяч единиц при норме в сотню.
Но опять же, из-за низкой осведомленности о болезни Дюшенна, врачи ошибочно диагностируют гепатит у таких мальчиков. Все дело в том, что при этом этом заболевании в крови повышаются уровни трансаминаз, что также происходит при гепатите. Но при дистрофии Дюшенна эти ферменты мышечного происхождения, а не печеночного.
Поставить точный диагноз можно только с помощью специализированного генетического анализа, направить на который должен врач. Бесплатную диагностику проводит в ФГБНУ «Медико-генетический научный центр имени академика Н.П. Бочкова» при значениях КФК более 2000 ЕД\л.
Чтобы направить пациента на диагностику, специалист должен позвонить на горячую линию для врачей по телефону 8-800-100-17-60 и зарегистрировать ребенка в программе бесплатного генотипирования.