The muscular system performs a number of important functions in the human body; motor abilities and coordination of movements directly depend on its proper functioning. Pathological processes accompanied by damage to muscle structures are extremely difficult for a person to tolerate and in most cases lead to disability. One of the most severe pathologies of this nature is Duchenne myopathy.
In Vivantes clinics, only diagnosis of this disease is possible.
In Germany, diagnosis and treatment of Duchenne myopathy is carried out by qualified doctors of the highest category. Specialists conduct a full range of studies to verify the disease. Based on the diagnostic results, treatment tactics are individually developed, which are based on innovative methods of conservative therapy.
What is Duchenne myopathy
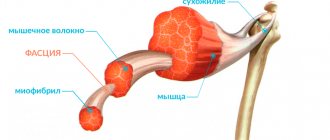
Myopathy or progressive Duchenne muscular dystrophy is a pathological process characterized by damage to the muscular structures of the body and leading to muscle weakness. One of the main features of the disease is the fact of constant, gradual progression. The disease debuts and is detected in the first years of a person’s life, mainly in children aged 2 to 5 years.
A characteristic feature of Duchenne myopathy is the fact that this disease is transmitted genetically. The hereditary pathology is linked to the X chromosome, that is, it is transmitted to the child from the mother. At the same time, women from whom children inherit the disease never get it.
The pathological process is accompanied by degenerative damage to muscle structures. Duchenne myopathy is characterized not only by its early onset, but the disease is prone to rapid progression. As a result, the child develops muscle weakness, the consequences of which are various skeletal deformities. In the later stages of progression, damage to the heart muscle and concomitant heart failure occur.
The true cause of Duchenne muscular dystrophy is an inherited genetic mutation. In this case, mutations are divided into two groups:
- In 70% of cases, the cause is a defect in the dystrophin gene inherited from the mother;
- In the remaining 30% of cases, the development of Duchenne disease is caused by local mutations in the mother's eggs.
The severe course of the pathological process is mainly due to the complete cessation of dystrophin synthesis. As a result, the muscles lose their contractility and are susceptible to a decrease in tone and overall strength.
Literature
- van Putten M., Hulsker M., Nadarajah VD, van Heiningen SH, van Huizen E., van Iterson M. et al. (2012). The effects of low levels of dystrophin on mouse muscle function and pathology. PLoS One. 7 , e31937;
- Russo FB, Cugola FR, Fernandes IR, Pignatari GC, Beltrão-Braga PC (2015). Induced pluripotent stem cells for modeling neurological disorders. World J. Transplant. 5, 209–221;
- Falzarano M. S., Scotton C., Passarelli C., Ferlini A. (2015). Duchenne muscular dystrophy: from diagnosis to therapy. Molecules. 20, 18168–18184;
- Bushby K., Finkel R., Birnkrant DJ, Case LE, Clemens PR, Cripe L. et al. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9, 77–93;
- Bushby K., Finkel R., Birnkrant DJ, Case LE, Clemens PR, Cripe L. et al. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 9, 177–189;
- Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E. et al. (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 351, 400–403;
- Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM et al. (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 351, 403–407;
- Tabebordbar M, Zhu K, Cheng JK, Chew WL, Widrick JJ, Yan WX et al. (2016). In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 351, 407–411;
- Elements: “The prokaryotic immune system will help edit the genome”;
- Gallagher J. (2016). Scientists get 'gene editing' go-ahead. BBC News;
- CRISPR systems: immunization of prokaryotes;
- Mutagenic chain reaction: genome editing on the verge of science fiction;
- Shouldn't we try to… change the genome?;
- Nobel Prize in Physiology or Medicine (2012): induced stem cells;
- Alzheimer's disease: the gene I'm crazy about;
- How to save Thirteen? (Prospects for the treatment of Huntington's disease);
- McGreevy JW, Hakim CH, McIntosh MA, Duan D. (2015). Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis. Model. Mech. 8, 195–213;
- Singh S. M., Kongari N., Cabello-Villegas J., Mallela K. M. (2010). Missense mutations in dystrophin that trigger muscular dystrophy decrease protein stability and lead to cross-β aggregates. Proc. Natl. Acad. Sci. USA. 107, 15069–15074;
- Goyenvalle A., Seto J. T., Davies K. E., Chamberlain J. (2011). Therapeutic approaches to muscular dystrophy. Hum. Mol. Genet. 20 , R69–R78.
Symptoms and diagnosis
The clinical picture of Duchenne myopathy becomes increasingly varied as the disease progresses. In this case, muscle weakness begins in the lower extremities, gradually spreading upward to the hips and pelvic muscles. Due to the fact that the pathology debuts at an early age, the first signs are expressed in specific developmental delays in the child. the following main symptoms of Duchenne disease :
- gait disturbances in children, with the child starting to crawl and walk late;
- when trying to stand up, the child clearly leans on his hands or gets up from his knees;
- formation of a “wasp” waist due to muscle atrophy;
- gradual loss of tendon reflexes;
- there are various forms of curvature of the spinal column (kyphosis, lordosis);
- deformation of the chest (for example, keeled structure);
- disorders of the cardiovascular system;
- as a rule, by the age of 12 the patient loses the ability to walk, and by the age of 15 he can independently perform most other movements;
- Various forms of mental retardation, including autism and other disorders, are also present in the clinical picture.
When starting a diagnosis, doctors attach special importance to recording all existing symptoms, studying the medical history, and a general examination of the patient. In addition, laboratory and instrumental studies are carried out, among which the most relevant are:
- detailed blood test;
- general clinical examination of urine;
- biochemical blood test;
- X-ray examinations;
- Ultrasound of the abdominal organs, retroperitoneum, heart;
- echocardiography;
- study to determine the concentration of dystrophin in muscle tissue;
- muscle biopsy;
- consultations with specialized specialists (cardiologist, geneticist).
"Amondis 45": safety of casimersen in the treatment of Duchenne muscular dystrophy
Studies in male mice and rats have demonstrated renal toxicity of casimersen, manifested by renal tubular degeneration. Although human trials have not reported anything similar, it is important to consider that some other antisense oligonucleotides have been studied in patients who have experienced renal toxicity, including potentially fatal glomerulonephritis. Therefore, therapy with Amondis 45 should be accompanied by monitoring of renal function.
The most common adverse reactions to casimersen were: upper respiratory tract infections (65% of patients versus 55% in the placebo group), cough (33% versus 26%), fever (33% versus 23%), headache (32). % vs. 19%), joint pain (21% vs. 10%), pain in the oropharynx (21% vs. 7%).
Treatment of Duchenne myopathy
The basic principles of treatment of Duchenne myopathy are based on the use of conservative therapy, medications and physiotherapeutic procedures. A treatment plan for each patient is developed individually. In this case, specialists take into account the presenting symptoms, diagnostic results, the patient’s age, and the degree of progression of muscular dystrophy.
The main goal of doctors in the treatment of Duchenne myopathy is to maximally slow down the progression of dystrophic processes and improve the patient’s quality of life. As part of complex drug therapy, the main groups of drugs are:
- Glucocorticosteroids - the action of these drugs is aimed at slowing the development of the disease; in the early stages, the latest generation drugs used can even stop the development of the pathology for some time.
- Prozerin is a specialized drug that helps improve the transmission of nerve impulses in muscle structures, thereby maintaining muscle tone.
- Calcium supplements – ensure the maintenance of normal muscle fiber contraction.
- Vitamin complexes are used primarily to improve metabolic processes, which also helps maintain muscle tone.
As for physiotherapy, specialists in Germany individually develop exercise therapy and massage programs and prescribe the wearing of special support splints. Taking into account the uniqueness of each clinical case, specialists take all possible measures to help the patient, improve quality and maximize life expectancy, limiting the progression of the disease.
In Vivantes clinics, only diagnosis of this disease is possible.
results
In 63% of patients with DMD, the disease debuted at the age of 2-5 years, in 37% - at 5-7 years. In all patients, the disease began with damage to the muscles of the pelvic girdle and proximal legs and was steadily progressive. The main clinical symptoms were weakness and atrophy of the muscles of the legs and pelvic girdle with the development of pseudohypertrophy of the calf muscles (Fig. 1).
Rice. 1. Patient S., 5 years old. Diagnosis of DMD, deletion in exon 52, pseudohypertrophy of the gastrocnemius muscles. Contractures in the knee and ankle joints were detected, and some patients had severe spinal deformities (Fig. 2).
Rice. 2. Patient R., 15 years old. Diagnosis of DMD, deletion in exon 44-50, severe kyphoscoliosis. Clinical symptoms of cardiopathy were observed in 68% of patients, pneumopathy - in 53%, 13% of patients were on mechanical ventilation at the time of the examination. A neuropsychological study revealed severe cognitive impairment in 33% of cases, and moderate cognitive impairment in 19% of cases.
Data from an enzyme immunoassay of neurotrophins indicate that NGF concentrations in the blood serum in patients with DMD are higher than in the control group - 2391 [1587; 4136] and 553 pg/ml [314; 864] respectively ( p
<0,001).
A molecular genetic study revealed the presence of deletions and duplications in 58% of cases, and nonsense mutations in various exons in 8% of cases. The distribution of mutations in the DMD gene was uneven; they were most often found in the gene region from exons 43 to 50 (Fig. 3).
Rice. 3. Distribution of patients by mutations in the DMD gene by exon. The x-axis is exons, the y-axis is the number of patients.
Analysis of the location of the mutation in the DMD gene revealed that there is impaired synthesis of dystrophin protein isoforms Dp260 ( n
=17), isoforms Dp140 (
n
=15), Dp116 (
n
=1) and Dp71 (
n
=1).
In 15 patients, there was a mutation in the DMD gene in exons affecting the synthesis of two (Dp260 and Dp140) ( n
= 14) or more isoforms (Dp140, Dp260, Dp116 and Dp71) (
n
= 1) (Table 1).
Table 1. Basic molecular genetic and clinical characteristics of the examined DMD patients Note.
The 5'UTR designation means that the 5' untranslated region of the Dp140 gene, which does not code for a protein but contains regulatory elements, is damaged. Due to the fact that the 5'UTR is very long (about 1000 kb), not all mutations within it may have the same damaging effect. The results of clinical neurological, molecular genetic and laboratory studies were compared in order to clarify the role of genetic and biochemical factors in the development of cognitive disorders in DMD. A significant decrease in the concentration of the neurotrophin BDNF was detected in group 2. Checking the empirical distribution of variables characterizing the concentration of neurotrophins in accordance with the law of normal distribution using the Shapiro-Wilk test showed a significant difference ( p
<0.01). Thus, to describe the numerical characteristics of these quantitative characteristics, it is most adequate to use the median and quartiles: Me [Q25%; Q75%], here and below the average tendency of a quantitative random variable will be described by the median, and the features of the distribution law will be specified by the boundaries of the interquartile range (Fig. 4).
Rice.
4. Concentration of BDNF, pg/ml (ordinate axis), in blood serum in the study groups (x axis). Thus, the concentration of BDNF has statistically significant differences in the groups of patients with and without cognitive disorders ( p
<0.001). In group 2, a reduced concentration of BDNF was observed - 23,670 [21,700; 30,720] pg/ml versus 32,700 [31,660; 33,750] pg/ml in group 1. For this indicator, the absolute risk of the variant of DMD with cognitive impairment, the odds ratio of this risk (OR) and the 95% confidence interval (CI) of the OR were calculated. The DFG level was divided into two intervals in accordance with the law of distribution of this variable in groups. In table 2 presented
Table 2. Estimation of the absolute risk of developing cognitive impairment in DMD depending on the concentration of BDNF Note.
* — comparison group or minimal predicted risk. results of determining the absolute risk of DMD with the development of cognitive impairment and the OR of this risk when assessing the relationship between the concentration of BDNF and the course of DMD. Based on the analysis of the distribution law, it was revealed that the minimal risk of DMD with the development of cognitive impairment, amounting to 20% (2 out of 10 patients), was observed in patients with a BDNF concentration of more than 31,000 pg/ml. Compared with this group, in patients with BDNF concentrations less than 31,000 pg/ml, the risk of an unfavorable course was 75% (15 out of 20 patients), the chances of developing cognitive disorders were statistically significant ( p
<0.001) increased more than 10 times (OR=12.0; 95% CI OR=[1.9–76.4]).
Neurotrophins NGF and CNTP did not have significant differences between the concentration indicators in the 1st and 2nd groups. When studying the concentration of neurotrophins depending on the location of the mutation in the DMD gene, a significant decrease in the concentration of NGF was revealed in the group with a distal location of the mutation. To clarify the role of the location of the mutation in the DMD gene and the development of cognitive disorders in patients, a comparison of data from molecular genetic and clinical neuropsychological studies was carried out, which did not reveal significant differences in the presence of cognitive disorders and the location of the mutation in the DMD gene. However, more pronounced cognitive impairment was observed in patients with DMD with a mutation located in the distal region of the DMD gene. The study of the role of individual dystrophin protein isoforms in the development of cognitive disorders did not reveal reliable data due to insufficient clinical observations.
Prof. Dr. Bettina Schmitz
Neurology – Epilepsy
Head of the Department of Neurology
Specialization
- Epilepsy
- Differential diagnosis of non-epileptic paroxysms
- Long-term EEG - video monitoring
- Medical and non-medical care for paroxysms and complications associated with epilepsy
- Member of the British Medical Association
- Instructor and member of the European Academy for the Study of Epilepsy (EUREPA)
- Member of the Commission on Psychobiology and the International League Against Epilepsy (ILAE)
- Psychoorganic syndrome, World Federation of Societies of Biological Psychiatry (WFSBP)
- Video consultation
Show doctor's personal profile
Discussion
A clinical and neuropsychological study of patients with DMD revealed cognitive impairment of varying severity in 33% of cases. In recent years, the study of the pathogenesis of cognitive disorders in patients with DMD has had a priority - identifying the connection between cognitive deficits and the location of the mutation in the DMD gene. It was noted that among patients with DMD with deletion of exon 52, cognitive disorders were more often diagnosed [21]. However, a number of researchers [22] explain this by the fact that in DMD, mutations in exons 44–53 are more often detected, thereby forming a larger cohort of patients. Among the patients we examined, 65% had mutations in the DMD gene located in exons 43-53, i.e., the distal part of the gene. It is known [23] that the distal location of the mutation in the DMD protein gene is a predisposing factor for the development of cognitive disorders. In our study, more pronounced cognitive impairment was also recorded in patients with a distal location of the mutation in the DMD protein gene. The contribution of various DMD protein isoforms to the development of cognitive impairment is currently being actively studied [24]. It has been established [25, 26] that dystrophin isoforms Dp140 and Dp71 are especially important for cognitive disorders. According to our data, among the identified mutations, those affecting the synthesis of the Dp140 isoform predominated, since it is known that the promoter of the Dp140 isoform is located in intron 44. It should be noted that among 4 patients with DMD with severe cognitive impairment, 3 had a mutation affecting the Dp260 and Dp140, and in 1 the synthesis of isoforms Dp140, Dp260, Dp116 and Dp71 is damaged. These data show that more severe cognitive impairment is present in patients with a mutation in the DMD gene, which affects the synthesis of several isoforms of the DMD protein, but the small number of clinical observations does not allow us to assess the reliability of the results obtained. For the first time, an attempt was made to evaluate the effect of the concentration of neurotrophins in the blood serum of patients with DMD on the development of cognitive disorders and to compare the data obtained with the results of molecular genetic analysis. It was revealed that in the group of patients with DMD with the presence of cognitive disorders, there was a significant decrease in the concentration of the neurotrophin BDNF. Comparing our own observations with the data of N. Doorenweerd et al. [27] studied the microstructure of the brain in patients with DMD using quantitative magnetic resonance imaging, we can hypothetically explain the role of this factor in the development of cognitive deficit. It was noted that patients with DMD with the presence of a mutation affecting the Dp140 (–) isoform, and who underwent neuropsychological examination with worse results, had a smaller brain volume, as well as gray matter, compared to control groups and patients with Dp140 (+) DMD. who received a positive assessment during testing. The same authors, studying cerebral blood flow in patients with DMD, found [28] that some patients have reduced cerebral perfusion. However, these findings did not correlate with reductions in brain and gray matter volume in patients with DMD. At the same time, patients with cerebral hemodynamic disorders had mutations in the DMD gene affecting the Dp140 isoform. It has been suggested that the Dp140 isoform influences the functioning of vascular endothelial cells and astrocytes, indirectly affecting the state of cerebral perfusion. Probably, a decrease in brain volume can be hypothetically associated with a low level of the neurotrophin BDNF, since it is this factor that promotes the growth and differentiation of brain neurons [19]. We found that in patients with BDNF concentrations less than 31,000 pg/ml, the risk of an unfavorable course was 75% (15 out of 20 patients), the chances of developing cognitive disorders were statistically significant ( p
<0.001) increased more than 10 times (OR=12.0; 95% CI OR=[1.9–76.4]). Note that patients with DMD with a distally located mutation in the gene had a higher concentration of the neurotrophin NGF. According to the results of previous studies of neurotrophic regulation in children with severe forms of hereditary and acquired pathology of the central nervous system [29, 30], NGF is a marker of the pathological process. It is also necessary to take into account the neuritinhibitory effect of high concentrations of NGF, revealed in a series of experiments with the blood serum of patients with spinal muscular atrophy in an organotypic culture of nervous tissue [31, 32].
Thus, a number of mechanisms are involved in the development of cognitive impairment in patients with DMD, the combination of which leads to a more pronounced cognitive defect. Among them, both molecular genetic factors are noted - the distal location of the mutation in the DMD gene, the influence of various combinations of impaired synthesis of DMD protein isoforms (Dp140, Dp116, Dp260 and Dp71), and biochemical factors associated with the characteristics of neurotrophic regulation, manifested by a reduced level of the neurotrophin BDNF in the blood serum of patients with DMD and elevated NGF concentrations.
The authors declare no conflict of interest.
Prof. Dr. Jörg Müller
Neurology – Parkinson's, MS
Head of the Department of Neurology
Specialization
- Recognized international expert in Parkinson's disease, dystonia and tremor
- Neuromuscular diseases
- Multiple sclerosis
- Treatment using butolotoxin (dystonia, spasticity)
- Deep brain stimulation treatment
- Author of more than 70 scientific publications, is a member of international expert councils
Show doctor's personal profile
“Traditional” treatment methods can aggravate the disease
If the diagnosis is confirmed, the doctor will prescribe glucocorticosteroids - today this is the gold standard of treatment to slow the progression of the disease. Special physical therapy and comprehensive rehabilitation at different ages are also indicated. In 2020, the first drug for the treatment of Duchenne muscular dystrophy, Ataluren, was registered in Russia, but so far little is known about its effectiveness.
The creators of the educational project Red Balloons for parents of children with Duchenne disease warn about the dangers of various “treatment” methods from the Internet, which desperate mothers and fathers are clutching at like straws. Unfortunately, they not only provide no benefit, but may even accelerate the progression of the disease.
What you should never do:
- give excessive physical activity (engage in aggressive sports, climb and descend steep stairs, jump on trampolines);
- steam your feet, wash with very hot water, go to saunas and steam baths;
- do massage without first consulting a doctor;
- resort to “treatment” methods from the Internet: stem cells, leeches and body wraps do not help.
Prof. Dr. Bruno-Marcel Mackert
Neurology
Head of the Department of Neurology
Specialization
- Internationally recognized specialist in stroke, peripheral nervous system diseases and clinical electrophysiology
- Author of over 40 original publications
- Board of Directors of the Berlin Stroke Society (BSA)
- Member of the Board of Directors of the Berlin Center for Stroke Research (CSB)
Show doctor's personal profile
Prof. Dr. Jörg Wissel, MD, FRCP
Neurological rehabilitation
Head of the Department of Neurological Rehabilitation and Physiotherapy, Department of Neurology and Rehabilitation Center
Specialization
- Neurological rehabilitation after stroke, traumatic brain injury and spinal cord injury
- Rehabilitation therapy for Parkinson's disease and dystonia
- Botulinum therapy
- Treatment of spasticity
- Neurological rehabilitation using intrathecal baclofen therapy and deep brain stimulation
- Author of more than 90 scientific publications, member of several expert commissions
Show doctor's personal profile
This disease is often confused with hepatitis
If there is any doubt about the boy's health, the doctor should order a blood test for creatine phosphokinase (CPK) activity. This is an enzyme contained in skeletal muscles, a marker of their breakdown. In muscular dystrophies, including Becker's disease, it is released in enormous quantities - several tens of thousands of units against a normal rate of one hundred.
But again, due to low awareness about Duchenne disease, doctors misdiagnose hepatitis in such boys. The thing is that with this disease, the levels of transaminases in the blood increase, which also happens with hepatitis. But in Duchenne dystrophy, these enzymes are of muscle origin, not liver.
An accurate diagnosis can only be made with the help of a specialized genetic analysis, which should be referred by a doctor. Free diagnostics are carried out at the Federal State Budgetary Institution “Medical Genetic Research Center named after Academician N.P. Bochkova" with CPK values of more than 2000 IU/l.
To refer a patient for diagnosis, a specialist must call the hotline for doctors at 8-800-100-17-60 and register the child in the free genotyping program.