Диагностика
Если у пациента отсутствуют основные симптомы пурпуры (кровотечения и кровоизлияния), у врачей возникают сложности с установкой правильного диагноза. Особенно нужно быть внимательными, чтобы не перепутать пурпуру с сосудистыми аномалиями кожи.
Пурпуру диагностируют, как правило, по данным клинической и гематологической картины. Сначала врач-гематолог проводит осмотр пациента. Для подтверждения диагноза врач назначает больному пройти такие аппаратные осмотры и сдать анализы:
- общий клинический и биохимический анализы крови;
- исследование мочевины;
- общий клинический анализ мочи;
- миелограмма.
Также для установления пурпуры проводится и дифференциальная диагностика посредством выявления симптомов таких заболеваний, как: гемолитико-уремический синдром, пурпура гемолитическая микроангиопатическая и гепаторенальный синдром.
Как правило, пурпура лечится, но бывают случаи и летального исхода, если кровотечения и кровоизлияния очень обильные.
Пурпура Шенлейна—Геноха
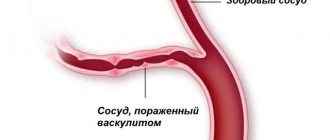
Пурпура Шенлейна—Геноха (геморрагический васкулит) — системный васкулит, поражающий микроциркуляторное русло (артериолы, капилляры и посткапиллярные венулы), с типичным отложением в стенке сосудов иммунных комплексов, состоящих из иммуноглобулинов А (IgA). Клинически болезнь проявляется кожной геморрагической сыпью, суставным синдромом, поражением желудочно-кишечного тракта (ЖКТ) и почек.
Пурпура Шенлейна—Геноха развивается в любом возрасте, однако максимальная заболеваемость наблюдается у детей в возрасте 4—6?лет, составляя примерно 13—18 случаев на 100 тыс. С возрастом заболеваемость снижается и развитие болезни после 60 лет считается редкостью.
Этиология заболевания не установлена, однако увеличение частоты развития пурпуры Шенлейна—Геноха в холодное время года, а также нередкая связь дебюта болезни с эпизодами острой респираторной или кишечной инфекции могут косвенно свидетельствовать об инфекционной природе заболевания. В список этиологических агентов, ассоциирующихся с развитием пурпуры Шенлейна—Геноха, входят ?-гемолитический стрептококк группы А, гемофильная палочка, хламидии, микоплазмы, легионеллы, иерсинии, вирусы Эпштейна—Барр, Коксаки, гепатита В и С, аденовирус, цитомегаловирус, парвовирус В19, сальмонеллы, Helicobacter pylori, Clostridium difficile. Имеются единичные наблюдения пурпуры Шенлейна—Геноха, развившейся после проведения вакцинации против брюшного тифа, кори, гриппа. В качестве триггеров заболевания могут выступать алкоголь, лекарства, пищевые продукты, переохлаждение, укусы насекомых.
Патогенез
В настоящее время пурпура Шенлейна—Геноха рассматривается как иммунокомплексное заболевание, связанное с отложением в сосудистой стенке гранулярных IgA-депозитов и активацией комплемента. Эта концепция основывается на результатах многочисленных исследований, показавших нарушение синтеза и/или метаболизма IgA у большинства больных пурпурой Шенлейна—Геноха: повышение уровня сывороточного IgA, IgA-содержащих иммунных, а также IgA-фибронектиновых комплексов. Тем не менее патогенетическое значение этих нарушений требует дальнейшей оценки. В последние годы накапливаются факты, свидетельствующие о противовоспалительных свойствах IgA, что дает основание расценивать увеличение его синтеза как компенсаторный процесс, возникающий вторично в ответ на воспалительную реакцию в слизистых оболочках. Так, было показано, что IgA обладает способностью уменьшать продукцию провоспалительных цитокинов и не способен активировать комплемент; IgA обнаруживается в эндотелии непораженных сосудов и в мезангии неизмененных почечных клубочков; описано наблюдение пурпуры Шенлейна—Геноха при полном селективном дефиците IgA. Учитывая нередкую связь развития пурпуры Шенлейна—Геноха с эпизодами инфекций респираторного тракта и ЖКТ, это предположение представляется вполне вероятным.
Другой причиной изменений в метаболизме IgA при пурпуре Шенлейна—Геноха может быть нарушение О-гликозилирования шарнирной области тяжелых цепей молекулы IgA1, что, как было показано, может приводить к нарушению клиренса IgA1 рецепторами печени и удлинению периода циркуляции IgA-полимеров и IgA-содержащих иммунных комплексов в системном кровотоке. Показано, что молекулы IgA1 с аберрантным гликозилированием приобретают способность активировать комплемент по альтернативному пути и имеют повышенную тропность к мезангиальному матриксу почечных клубочков.
В последние годы получены дополнительные данные, косвенно подтверждающие предположение об инфекционно зависимом характере пурпуры Шенлейна—Геноха. Так, было показано, что у большинства больных в период обострения кожного васкулита наблюдается транзиторная эндотоксемия — циркуляция в системном кровотоке липополисахарида грамотрицательных бактерий. Патогенетическое значение этого феномена при пурпуре Шенлейна—Геноха требует дальнейшего изучения, однако предполагается возможность участия эндотоксина в развитии сосудистого воспаления, опосредованного реакцией Шварцмана. Важную роль в патогенезе эндотоксемии может играть хроническое воспаление кишечной стенки, возможно, обусловленное дисфункцией ее локальной иммунной системы или инфекционным процессом. В пользу этого предположения свидетельствует обнаружение повышенной кишечной проницаемости для макромолекул (овальбумин) у большинства больных пурпурой Шенлейна—Геноха в период обострений кожного васкулита. Кроме этого продемонстрировано наличие у больных пурпурой Шенлейна—Геноха хронического воспалительного процесса в слизистой оболочке тонкой кишки, что, по-видимому, является морфологической основой для нарушения функции кишечного барьера и развития транзиторной эндотоксемии.
Клиническая картина
Клиническая картина пурпуры Шенлейна—Геноха складывается из четырех типичных проявлений: кожной геморрагической сыпи, поражения суставов, ЖКТ и почек. В большинстве случаев болезнь развивается исподволь, постепенно и существенно не нарушает общее состояние больных. Как правило, такой вариант начала болезни наблюдается при изолированном поражении кожи. Количество органных проявлений пурпуры Шенлейна—Геноха варьирует от 1—2 до комбинации всех 4 классических признаков, которые могут развиваться в любой последовательности в течение нескольких дней или недель болезни. В некоторых случаях, кроме упомянутых проявлений, может развиваться поражение других органов, в частности, легких, сердца, центральной нервной системы.
Поражение кожи наблюдается у всех больных пурпурой Шенлейна—Геноха и является обязательным критерием диагностики. В большинстве случаев геморрагическая сыпь является первым клиническим проявлением болезни, к которому в дальнейшем присоединяется поражение других органов и систем. Наиболее типичная локализация кожных высыпаний: нижние конечности — голени и стопы. Нередко кожная сыпь распространяется на бедра, ягодицы, туловище, верхние конечности и исключительно редко на лицо. В процессе эволюции геморрагии постепенно бледнеют, трансформируются в коричневые пигментные пятна и затем исчезают. При длительном рецидивирующем течении кожа в области поражения может пигментироваться вследствие развития гемосидероза. В большинстве случаев геморрагическая сыпь представлена петехиями и пурпурой, однако в ряде случаев могут также наблюдаться эритематозно-макулярные и уртикарные элементы.
Поражение суставов, как правило, развивается параллельно с поражением кожи и протекает по типу мигрирующих полиартралгий, реже — артритов. Излюбленная локализация — коленные и голеностопные суставы, реже поражаются локтевые, лучезапястные и другие суставы. Эти проявления болезни всегда преходящи и доброкачественны, никогда не приводят к развитию стойких изменений в суставах.
Поражение ЖКТ отмечается у 60—80% больных детского возраста и у 40—65% взрослых больных. Наиболее постоянный симптом: боли в животе, усиливающиеся после еды, что нередко создает типичную картину «брюшной жабы». Частым осложнением абдоминального поражения при пурпуре Шенлейна—Геноха является кишечное кровотечение.
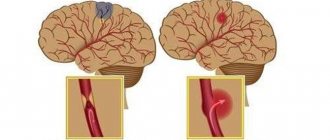
Поражение почек при пурпуре Шенлейна—Геноха может приобретать хроническое течение и является главным фактором, определяющим прогноз болезни в целом. Частота вовлечения почек варьирует от 30 до 70% в зависимости от возраста больных. У взрослых поражение почек выявляется почти в 2 раза чаще, чем у детей. Как правило, клинические признаки поражения почек выявляют в первые 3 месяца заболевания, тем не менее при хроническом рецидивирующем течении кожного васкулита возможно отсроченное возникновение признаков гломерулонефрита — спустя несколько месяцев или даже лет после дебюта болезни. Возможными предвестниками вовлечения в процесс почек у детей являются мужской пол, возраст старше 5 лет, абдоминальный синдром, персистирующая кожная пурпура и снижение уровня фактора XIII в плазме крови. У взрослых больных к факторам риска поражения почек относят наличие лихорадки и эпизоды инфекций в дебюте болезни, распространение кожной сыпи на туловище, тяжелый абдоминальный синдром и наличие лабораторных признаков воспалительной активности болезни. Тяжесть почечной патологии, как правило, не коррелирует с выраженностью кожных проявлений болезни, однако как у детей, так и у взрослых отмечена достоверная положительная корреляция между частотой поражения почек и развитием абдоминального синдрома, что требует более тщательного динамического наблюдения за соответствующим контингентом больных. У детей в половине случаев поражение почек имеет благоприятное течение с полным клинико-лабораторным выздоровлением, в то время как у большинства взрослых больных наблюдается тенденция к хроническому персистирующему течению нефрита.
У половины больных пурпурой Шенлейна—Геноха гломерулонефрит проявляется микрогематурией, которая, как правило, сочетается с минимальной или умеренно выраженной протеинурией. У трети больных наблюдается макрогематурия, которая чаще всего развивается в дебюте нефрита, но может возникать и на более поздних этапах почечного поражения во время обострений кожного васкулита или респираторных инфекций. Возможны и более тяжелые варианты почечного поражения, в том числе нефротический синдром, быстропрогрессирующий нефрит и острая почечная недостаточность. У 14—20% больных обнаруживается синдром артериальной гипертензии. Развитие хронической почечной недостаточности (ХПН) в исходе гломерулонефрита отмечается у 12—30% больных.
Диагностика
Диагностика пурпуры Шенлейна—Геноха основывается на выявлении типичных клинических признаков заболевания, в первую очередь двусторонних кожных геморрагических высыпаний в момент осмотра или в анамнезе. Специфических лабораторных тестов при пурпуре Шенлейна—Геноха не существует. Изменения в клиническом анализе крови — повышение скорости оседания эритроцитов (СОЭ) — могут отражать воспалительную активность болезни, а также выраженность осложнений (анемия при кишечном кровотечении). Наличие тромбоцитопении — критерий исключения пурпуры Шенлейна—Геноха. Выраженное повышение СОЭ и значительная диспротеинемия не характерны для пурпуры Шенлейна—Геноха. Активность болезни отражают уровень фактора Виллебранда и тромбомодулина в плазме крови. Обнаружение высокого уровня продуктов деградации фибрина/фибриногена в плазме при активных формах болезни не является признаком развития ДВС-синдрома, а лишь отражает высокую воспалительную активность заболевания. В план обследования всех больных необходимо включать вирусологическое и иммунологическое исследование крови для исключения других заболеваний, протекающих с кожной пурпурой.
Ключевую роль в подтверждении клинического диагноза играет биопсия кожи и/или почек, реже других органов, с обязательным проведением иммуногистохимического исследования, выявляющего фиксацию в сосудистой стенке IgA-содержащих иммунных комплексов. Следует учитывать, что кроме пурпуры Шенлейна—Геноха, IgA-депозиты обнаруживают при поражении кожи в рамках хронических воспалительных заболеваний кишечника (болезнь Крона, язвенный колит), хронических диффузных заболеваний печени алкогольной этиологии, целиакии, герпетиформного дерматита Дюринга.
Морфологическая картина поражения почек при пурпуре Шенлейна—Геноха идентична таковой при болезни Берже (первичной IgA нефропатии). Наиболее частый морфологический вариант поражения почек — мезангиопролиферативный гломерулонефрит, характеризующийся фокальной или диффузной пролиферацией мезангиоцитов. Иммуногистохимическое исследование выявляет гранулярные депозиты IgA, реже IgG, а также С3-компонента комплемента, фибрина. В более тяжелых случаях отмечается формирование эпителиальных «полулуний».
Широко используемые классификационные диагностические критерии пурпуры Шенлейна—Геноха, предложенные в 1990?г. Американской коллегией ревматологов и включающие возраст больного менее 20 лет, пальпируемую пурпуру, абдоминальный синдром и морфологическую картину кожного лейкоцитокластического васкулита (необходимо наличие 2 и более из 4 критериев), имеют малую практическую значимость вследствие их низкой чувствительности и специфичности (87,1 и 87,7%, соответственно).
Дифференциальная диагностика
Дифференциальная диагностика проводится с широким кругом заболеваний, протекающих с поражением сосудов мелкого калибра:
- первичные васкулиты сосудов мелкого калибра (гранулематоз Вегенера, микроскопический полиангиит, синдром Черджа–Стросс, криоглобулинемический васкулит). Дифференциально-диагностическое значение имеют результаты исследования крови на антинейтрофильные цитоплазматические антитела (гранулематоз Вегенера, микроскопический полиангиит, синдром Черджа–Стросс) и криоглобулины (криоглобулинемический васкулит); данные морфологического исследования (гранулематозное воспаление при гранулематозе Вегенера, эозинофильный васкулит при синдроме Черджа—Стросс). Особое значение имеет иммуногистохимическое исследование биоптатов пораженных тканей. Обнаружение IgA-депозитов является характерным признаком пурпуры Шенлейна—Геноха.
- Васкулиты при аутоиммунных заболеваниях (системной красной волчанке, ревматоидном артрите, болезни Шегрена, болезни Крона, язвенном колите). Дифференциация основывается на клинических особенностях, свойственных каждой нозологии, результатах лабораторно-инструментальных методов исследования.
- Васкулиты при инфекциях (подострый инфекционный эндокардит, туберкулез, инфекция вирусами гепатита В и С), злокачественных новообразованиях, лекарственной аллергии.
Лечение
При поражении кожи могут быть эффективны такие лекарственные средства группы сульфаниламидов: сульфасалазин (внутрь по 500—1000 мг 2 раза в сутки), колхицин (внутрь, 1—2 мг 1 раз в сутки). Глюкокортикоиды эффективны у подавляющего числа больных, особенно в высоких дозах, однако их длительное применение у больных пурпурой Шенлейна—Геноха без вовлечения внутренних органов нежелательно, поскольку выраженность побочных эффектов в такой ситуации может превосходить тяжесть самой болезни. Назначение нестероидных противовоспалительных препаратов (НПВП) оправдано лишь при выраженном суставном синдроме и неэффективности других лекарственных средств. В остальных случаях от назначения НПВП следует воздерживаться вследствие неблагоприятного действия на слизистую оболочку кишечника и усиления кишечной проницаемости.
Поражение желудочно-кишечного тракта с интенсивными абдоминалгиями — абсолютное показание к назначению глюкокортикоидов:
преднизолон внутривенно капельно 300—500 мг/сут в течение 3 дней подряд с последующим переходом на прием внутрь 0,5 мг/кг 1 раз в сутки в течение 2—3 недель, затем быстрое снижение дозы по 5?мг каждые 3 суток до полной отмены.
Желудочно-кишечное кровотечение (в том случае, если оно обусловлено васкулитом, а не иными причинами) не является противопоказанием к назначению глюкокортикоидов внутрь, а, напротив, служит одним из основных показаний к такому лечению. Противопоказанием к назначению глюкокортикоидов внутрь при абдоминальном синдроме может быть только перфорация стенки кишки, которая в настоящее время крайне редко осложняет течение пурпуры Шенлейна—Геноха.
Наибольшие проблемы медикаментозного лечения пурпуры Шенлейна—Геноха связаны с выбором средств лечения хронического гломерулонефрита. Большинство авторов считают оправданным применение сверхвысоких доз глюкокортикоидов, цитостатиков и/или сеансов плазмафереза в случае тяжелого гломерулонефрита (нефротический синдром с нормальной или нарушенной функцией почек; более 50% клубочков с эпителиальными «полулуниями»).
В этом случае используется следующая схема:
- преднизолон внутрь 1 мг/кг 1 раз в сутки в течение 4—6 недель, затем снижение дозы по 2,5 мг/недель до полной отмены или преднизолон внутривенно капельно 15?мг/кг 1 раз в сутки в течение 3 дней подряд (всего 6—20 трехдневных «пульсов» с интервалом в 3—4 недели); +
- циклофосфамид внутривенно капельно 15 мг/кг 1 раз в 3—4 недели, под контролем уровня лейкоцитов периферической крови и трансаминаз (всего 6—20 «пульсов»); +
- плазмаферез с объемом эксфузии 30—60?мл/кг, 10—14 сеансов.
В единичных неконтролируемых исследованиях показана эффективность при тяжелых вариантах поражения почек комбинации глюкокортикоидов и азатиоприна, а также комбинации глюкокортикоидов и циклофосфамида с антиагрегантами или антикоагулянтами.
Кроме того, для лечения больных с нефротическим и быстропрогрессирующим гломерулонефритом предлагается использовать внутривенные иммуноглобулины:
иммуноглобулин человеческий нормальный внутривенно по 400—1000 мг/кг в течение 1—5 сут, повторные курсы 1 раз в месяц в течение 6 месяцев.
В отношении менее тяжелых форм гломерулонефрита единства мнений нет. При изолированной микрогематурии, минимальной протеинурии (до 0,5 г/сут) и сохранной функции почек, как правило, активного иммуносупрессивного лечения не требуется. При умеренной протеинурии (0,5—1 г/сут) показано назначение лекарственных средств, влияющих на неиммунные механизмы прогрессирования поражения почек: ингибиторы ангиотензинпревращающего фермента и/или антагонисты рецепторов ангиотензина II (в связи с их способностью уменьшать внутриклубочковую гипертензию и выраженность протеинурии), статины (при нарушении липидного обмена). В некоторых ретроспективных исследованиях показан благоприятный эффект тонзиллэктомии на течение нетяжелых форм гломерулонефрита при пурпуре Шенлейна—Геноха.
Коррекция нарушений гемостаза, ранее считавшаяся первоочередной задачей в лечении пурпуры Шенлейна—Геноха, в настоящее время рассматривается лишь в качестве вспомогательного метода терапии, перспективы которого оцениваются скептически. В практическом плане представляют интерес сообщения японских исследователей о благоприятном долгосрочном клиническом и патоморфологическом эффекте фибринолитической терапии урокиназой на течение гломерулонефрита при пурпуре Шенлейна—Геноха:
урокиназа внутривенно медленно 5000 МЕ/кг 3 раза в неделю в течение 3—12?недель.
По мнению авторов, действие урокиназы может быть основано на уменьшении выраженности внутриклубочковой гиперкоагуляции и растворении депозитов фибриногена/фибрина.
Прогноз
Важным клиническим прогностическим фактором, определяющим частоту развития ХПН, является выраженность протеинурии. Так, если при минимальной протеинурии ХПН развивается у 5% больных, то при нефротическом синдроме этот показатель повышается до 40—50%. Наиболее неблагоприятным в отношении развития ХПН является сочетание нефротического синдрома с артериальной гипертензией и нарушением функции почек в дебюте гломерулонефрита.
Наиболее важным морфологическим критерием для определения прогноза поражения почек считают долю почечных клубочков с «полулуниями» от общего числа клубочков. Так, по данным французских авторов, наблюдавших 151 больного от 1 года до 18 лет, при наличии «полулуний» больше, чем в 50% клубочков терминальная почечная недостаточность развилась у 37% больных, а еще у 18% гломерулонефрит имел хроническое прогрессирующее течение. С другой стороны, у 85% пациентов, достигших терминальной почечной недостаточности, «полулуния» имелись более чем в половине почечных клубочков. У 70% больных с полным выздоровлением или минимальными изменениями в анализах мочи «полулуний» в клубочках не было найдено.
Важно, что у большинства больных с поздним прогрессированием гломерулонефрита отсутствуют клинические признаки активности почечных и внепочечных поражений, что объясняют преимущественным влиянием на течение почечного поражения неиммунных механизмов прогрессирования. В связи с этим у всех больных пурпурой Шенлейна—Геноха с поражением почек крайне важным является тщательный контроль артериального давления и коррекция метаболических нарушений, в частности гиперурикемии и дислипидемии.
Лечение
Суть лечения пурпуры сводится к тому, чтобы было уменьшено количество продуцирования антитромбоцитарных антител и стали невозможными их связывания с тромбоцитами.
Если у больного происходят кровотечения, то врач назначает ему курс гемостатических препаратов, а также — аминокапроновую кислоту. Если кровотечения еще и маточные, то пациенткам приписывается окситоцин.В том случае, если есть риск кровоизлияния в головной мозг, врачи-хирурги назначают больному вливание (инфузию) тромбоцитов.
Существует также хирургический метод лечения пурпуры. В этом случае больному проводят операцию по удалению селезенки (спленэктомия). Но очень важно помнить, что хирургические вмешательства – это крайний случай, когда консервативная терапия оказывается неэффективной.
Еще один метод лечения пурпуры – это плазмафарез, или процедура по очистке плазмы крови от антител и токсинов посредством фильтрования плазмы крови на специальных аппаратах.
Также больному пурпурой сосудистого вида врачи советуют соблюдать гипоаллергенную диету.
Болезнь Шенлейн — Геноха
Кожный синдром встречается наиболее часто. При нем симметрично поражаются конечности, ягодицы, реже — туловище. Возникает папулезно-геморрагическая сыпь, иногда с волдырями. Высыпания однотипные, сначала имеют отчетливую воспалительную основу, в тяжелых случаях осложняются центральными некрозами и покрываются корочками, надолго оставляют пигментацию. При надавливании элементы сыпи не исчезают.
Суставной синдром возникает часто вместе с кожным или спустя несколько часов или дней после него в виде болей разной интенсивности в крупных суставах (коленных, локтевых, тазобедренных). Через несколько дней боль проходит, но при новой волне высыпаний может возникнуть опять. В ряде случаев суставное поражение бывает стойким и упорным, напоминает ревматоидный полиартрит.
Абдоминальный синдром чаще наблюдается в детском возрасте (у 54-72% больных), приблизительно у 1/3 он преобладает в клинической картине, в ряде случаев предшествует кожным изменениям, что очень затрудняет диагностику. Основной признак — сильные боли в животе, постоянные или схваткообразные, иногда настолько интенсивные, что больные не находят себе места в постели и в течение многих часов кричат. Боль обусловлена кровоизлияниями в стенку кишки. Эти кровоизлияния могут сочетаться с пропитыванием кровью кишечной стенки и слизистой оболочки, кровотечениями из нее и из участков некроза, кровавой рвотой, меленой (примесью крови в кале) или свежей кровью в кале, а также ложными позывами с частым стулом или, наоборот, с его задержкой. С самого начала определяются лихорадка, более или менее выраженный лейкоцитоз (увеличение количества лейкоцитов в крови). При обильных кровотечениях развиваются коллапс (обморочное состояние) и острая постгеморрагическая анемия. В некоторых случаях частая рвота приводит к большой потере жидкости и хлоридов. В коагулограмме определяются гипертромбоцитоз и гиперкоагуляция.
У значительной части больных абдоминальный синдром непродолжителен и проходит самостоятельно за 2-3 дня. Периоды сильной боли могут чередоваться с безболевыми промежутками, продолжающимися около 1-3 ч. Это помогает отличить абдоминальный синдром от острых хирургических заболеваний органов брюшной полости. Особенно трудна такая дифференцировка у больных без кожно-суставных проявлений и с симптомами раздражения брюшины. Чаще абдоминальный синдром имитирует острую кишечную непроходимость (инвагинацию), аппендицит, перекруты и кисты яичника, прободение язвы кишечника.
Сравнительная диагностика может вызывать для врача определенные сложности — это связано с тем, что сам геморрагический васкулит может стать причиной всех перечисленных хирургических заболеваний органов брюшной полости. Так, например, описано немало случаев инвагинации (внедрения одного участка кишки в другой) и непроходимости кишки в связи со сдавливанием или закрытием ее просвета гематомой (особенно у детей моложе 2 лет), некроза кишки и ее перфорации (образование сквозного дефекта), острого аппендицита и других осложнений, требовавших хирургического вмешательства. Трудности дифференциальной диагностики в подобной ситуации приводят к тому, что часть больных геморрагическим васкулитом подвергается необоснованным хирургическим вмешательствам.
У взрослых больных абдоминальный синдром наблюдается реже и в большинстве случаев не служит основанием для диагностической лапаротомии, редко осложняется кишечной непроходимостью и перитонитом (воспалением брюшины). В пожилом
2.Причины
Этиопатогенез геморрагического васкулита Шенляйна-Геноха (заметим попутно, что первую из двух немецких фамилий корректней писать через «я») остается дискутабельным, однако к настоящему времени все больше исследователей склоняется к аутоиммунной гипотезе. Источником споров является, в основном, разнообразие и разнородность триггерных (пусковых) факторов, способных спровоцировать атаку иммунной системы на ткани собственных сосудов. К таким факторам относятся респираторные инфекции, вакцинация, вирусные заболевания, прием некоторых лекарственных средств (антибиотики, нейролептики первого поколения), аллергические реакции, беременность, переохлаждения, цирроз печени, онкопроцессы и т.д.
Посетите нашу страницу Терапия
Когда стоит обратиться к врачу?
Если вас начали беспокоить представленные ниже проблемы, то в срочном порядке запишитесь на прием к специалисту:
- Внезапное высыпание сыпи в виде крошки в районах рук, ног, тазобедренной области и лица;
- Регулярные болезненные ощущения и дискомфорт в суставах, в частности это касается нижних конечностей тела;
- Отеки и припухлости;
- Появление синячков;
- Наличие кровяных выделений в моче;
- Резкий скачок вверх давления.
Идиопатическая тромбоцитопеническая пурпура: от Верльгофа до наших дней
Спонтанное кожное кровотечение, известное более 2500 лет, уже тогда, в греческом и римском периоде врачевания, называлось «пурпурой». В 1735 году Паул Верльгоф (ИТП называют также «болезнью Верльгофа») описал 16-летнюю девочку с носовым кровотечением и кровотечением слизистых, которые купировались применением лимонной кислоты. Он назвал это заболевание «Morbus Maculosus Haemorhhagicus». Но заметного прогресса в лечении больных с ИТП достигли позже: в 1916 году в Праге профессор Шлоффер удалил селезенку у женщины с этим заболеванием. После операции последовало значительное увеличение числа тромбоцитов. И до сих пор спленэктомия является одним из вариантов лечения пациентов при ИТП. Однако наиболее полная картина этой патологии, наше представление о механизмах ее возникновения и подходах к диагностике и терапии начали складываться лишь в последнее время.
Идиопатическая тромбоцитопеническая пурпура (ИТП), или первичная иммунная тромбоцитопения (ИТП), является аутоиммунным приобретенным заболеванием, которое характеризуется изолированной тромбоцитопенией числом тромбоцитов ниже 100х109/л. Оно может проявляться геморрагическим симптомом различной степени выраженности – от петехиальных кожных кровоизлияний до угрожающих жизни кровотечений. Болеют и дети, и взрослые. Этиология ИТП неизвестна. Поэтому и называется «идиопатическая». Среди пусковых факторов наибольшую группу составляют инфекции, беременность, а также прививки, стрессы.
Известно, что доминирующий механизм развития тромбоцитопении при ИТП обусловлен выработкой аутоантител к структурам мембраны тромбоцитов и их предшественников – мегакариоцитов, которые приводят к повышенному разрушению тромбоцитов фагоцитами, главным образом в селезенке, реже в печени, и недостаточной выработкой тромбоцитов в костном мозге. У пациентов с ИТП вырабатываются главным образом IgG аутоантитела против гликопротеинов GPIIb/IIIa или GPIb/IX поверхности тромбоцитов. Процесс формирования иммунной реакции на собственные тромбоциты – сложный, многоступенчатый, циклический. В нем принимают участие В-лимфоциты, Т-лимфоциты, NK-клетки, макрофаги. Помимо антителообразования, большую роль в патогенезе ИТП играют субпопуляции Т-лимфоцитов, развитие дисбаланса Т-клеточного звена иммунного ответа. Выявлена связь между ИТП и некоторыми генами-кандидатами, что указывает и на наличие генетической предрасположенности к ИТП.
С учетом сопутствующей патологии у пациента образуется определенный фенотип заболевания. Таким образом, патогенез ИТП связан с глубокими нарушениями иммунной системы. В связи с этим идиопатическая тромбоцитопеническая пурпура переименована на первичную иммунную тромбоцитопению с неизвестной этиологией. Соответственно, при всех других формах иммунная тромбоцитопения с известной этиологией будет симптомом других аутоиммунных заболеваний – системной красной волчанки (СКВ), антифосфолипидного синдрома (АФЛС), ревматоидного артрита (РА) и др.
В настоящее время продолжается поиск этиопатогенетических механизмов развития этой редкой патологии, которые могли бы стратифицировать пациентов на группы риска для индивидуализации лечебной тактики.
В литературе ИТП описывается как редкое, орфанное, заболевание. Надо сказать, что в медицинском мире не существует единого определения этой группы заболеваний. В одних странах орфанные патологии выделяют в зависимости от количества страдающих, в других – от доступности методов лечения, в третьих – к редким заболеваниям относят только хронические, угрожающие жизни.
В России своя история орфанных заболеваний. С того времени, как в нашей стране законодательно было принято определение «орфанные заболевания» (закон № 323 «Об основах охраны здоровья граждан в РФ» от 21.11.2011)1, а именно: редкими (орфанными) заболеваниями являются заболевания, которые имеют распространенность не более 10 случаев заболевания на 100 тысяч населения, все онкогематологические и многие гематологические заболевания стали считаться орфанными. Что касается ИТП, согласно Постановлению Правительства РФ № 403 от 26.04.20122 идиопатическая тромбоцитопеническая пурпура (D69.3) была включена в короткий список жизнеугрожающих и хронических прогрессирующих редких (орфанных) заболеваний, приводящих к сокращению продолжительности жизни граждан или их инвалидизации. В этот короткий список были включены и такие гематологические состояния, как пароксизмальная ночная гемоглобинурия (болезнь Маркиафавы-Микели), апластическая анемия, наследственные болезни нарушения метаболизма, гемолитико-уремический синдром и др.
Все эти законодательные решения привели к тому, что в ФГБУ «НМИЦ гематологии» в 2012 году было создано отделение – стационар орфанных заболеваний (руководитель: профессор Е.А. Лукина). Нозологический диапазон патологий, которыми занимается отделение, очень широк.
Лекарства, разработанные для лечения редких заболеваний, также называются орфанными препаратами и включены список дорогостоящих ЛС. Назначение орфанного статуса заболеваниям и любым лекарственным препаратам – социальный, политический вопрос во многих странах, как и в России. Поддержка исследований редких болезней со стороны правительств привела к прорывам в медицине, которые не могли бы быть достигнуты в рамках до этого существующей системы финансирования.
Статус орфанного заболевания для ИТП также открывал новые возможности для улучшения его диагностики и лечения современными методами, которые не могли бы быть осуществлены без него. Речь идет прежде всего о двух орфанных дорогостоящих препаратах агонистов тромбопоэтиновых рецепторов (аТПО) (ромиплостим от «Новартис» и элтромбопаг от «Амджен»). Лекарственное обеспечение осуществляется за счет бюджетов субъектов Российской Федерации.
Эпидемиология
Надо напомнить, что в нашей стране на популяционном уровне заболеваемость ИТП до 2014 года не была изучена. И информации для оценки особенности течения, эффективности и безопасности различных вариантов терапии больных ИТП было недостаточно. Для решения этих проблем под эгидой Национального гематологического общества (председатель Наблюдательного совета НГО – главный внештатный гематолог РФ, директор ФГБУ «НМИЦ гематологии» Минздрава России, академик РАН, профессор В.Г. Савченко) в начале декабря 2012 года был сформирован и начал функционировать «Регистр заболеваний системы крови».3 С 2014 года стартовала работа и в его подразделе «ИТП» – началось многоцентровое проспективное наблюдательное когортное исследование «Эпидемиологические и клинические характеристики ИТП у взрослых в России» (руководитель: А.Л. Меликян).
Согласно данным регистра Национального гематологического общества (НГО), заболеваемость взрослого населения ИТП в РФ среднем составляет 2,0 (1,6‒3,6) на 100 тысяч населения в год. ИТП не имеет географических особенностей. Мужчины болеют в 2‒3 раза реже женщин. Наибольшая доля пациентов (45,4%) находилась в возрастной группе от 18 до 40 лет, в группе от 41 до 60 лет – 26,0% и старше 60 лет – 28,6%. Таким образом, среди пациентов с ИТП 71,4% находятся в трудоспособном возрасте. Наибольшая частота встречаемости ИТП была зарегистрирована у женщин в фертильном возрасте 4. Полученные нами результаты вполне сопоставимы с данными регистров других европейских стран.
Диагностика
Главным клиническим проявлением ИТП является геморрагический синдром, и прогноз течения заболевания всецело зависит от степени его тяжести. Риск развития кровотечений у больных с ИТП оценивается по количеству тромбоцитов в анализе периферической крови. Согласно данным регистра, в 70,0% случаев количество тромбоцитов в дебюте заболевания составляет от 3 до 30х109/л, среди них у 35% выявляется критический уровень тромбоцитов (от 3 до 10х109/л) с риском развития спонтанных настораживающих, жизнеугрожающих кровотечений, что требует незамедлительного назначения лечения.
Геморрагический синдром проявляется в виде: кожных геморрагий – 77% случаев; кровоточивости слизистых полости рта – 39%; носового кровотечения – 31%; менометроррагии – 15% (среди женщин); желудочно-кишечного кровотечения – 7%; гематурии – 4%; внутримозгового кровотечения – 0,9%, других – 1% (кровоизлияния в сетчатку глаз, геморроидального кровотечения).4
Таким образом, около 1/3 пациентов на момент постановки диагноза имеют геморрагические проявления, соответствующие тяжелой форме ИТП (3‒4-й степени кровоточивости по классификации ВОЗ). ИТП – заболевание не генетическое, но обычно сопровождает пациента на протяжении всей его жизни и является неизлечимым. Течение заболевания осложняется еще тем, что у 60‒70% пациентов через 12 месяцев (хроническая фаза) заболевание приобретает хронический, рецидивирующий характер, вновь.
ИТП – заболевание не генетическое, но обычно сопровождает пациента на протяжении всей его жизни и является неизлечимым. Течение заболевания осложняется еще тем, что у 60‒70% пациентов через 12 месяцев (хроническая фаза) заболевание приобретает хронический, рецидивирующий характер, вновь появляется геморрагический синдром, требующий проведения противорецидивного курса терапии.
Диагноз «ИТП» – это диагноз исключения, т.е. до настоящего времени нет ни одного специфического теста заболевания. Тромбоцитопении различного генеза регистрируются при широком спектре заболеваний гематологической, негематологической и врожденной природы, у которых длительное время доминирующим клиническим симптомом может быть изолированная тромбоцитопения. Поэтому для установления истинных причин тромбоцитопении необходимо проведение расширенного диагностического поиска в дебюте заболевания.4
Первоначальный подход к диагностике причин тромбоцитопении основывается на анамнезе пациента (его фоновых заболеваниях и предшествующей медикаментозной терапии), его объективном физическом осмотре и обследовании по протоколу. Разработанный нами протокол дифференциального диагноза тромбоцитопений включен в Национальные клинические рекомендации по ИТП.5 Самое главное, что все предложенные лабораторные и инструментальные исследования существуют в рутинной практике и их проведение обязательно для всех пациентов с подозрением на ИТП.
После исключения других причин тромбоцитопении диагноз ИТП устанавливается на основании следующих критериев:
- изолированная тромбоцитопения менее 100,0х109/л, зарегистрированная минимум в двух последовательных анализах крови;
- отсутствие морфологических и функциональных аномалий тромбоцитов;
- отсутствие патологии лимфоцитов, гранулоцитов и эритроцитов;
- нормальные показатели гемоглобина, эритроцитов и ретикулоцитов, если не было существенной кровопотери;
- повышенное или нормальное количество мегакариоцитов в миелограмме;
- нормальные размеры селезенки.
Важно иметь в виду: нередко для быстрого купирования геморрагического синдрома пациентам без обследования по протоколу назначаются кортикостероиды, что смазывает истинную клиническую картину вторичных иммунных тромбоцитопений и влияет на истинные результаты иммунологических анализов. По данным нашего отделения, до 15‒20% случаев в динамике при повторном обследовании по протоколу диагноз ИТП заменяется на другой. Картина болезни может меняться с течением времени, в связи с этим необходимо постоянно обновлять данные о состоянии больного, дифференциальную диагностику проводить на каждом этапе наблюдения/терапии ИТП. Таким образом, очень важно дифференциальный диагноз проводить между первичной и вторичной тромбоцитопенией не только в дебюте заболевания, но и при рецидиве тромбоцитопении.
Установление истинных причин тромбоцитопении крайне важно для выбора адекватной терапии таких пациентов
Во многих случаях пациенты с первичной и вторичной ИТП получают аналогичное лечение. Тем не менее, если ИТП развивается на фоне основного заболевания (например, СКВ, АФС, HCV-инфекции, ВИЧ-инфекции или лимфопролиферативного заболевания), то лечение должно быть направлено главным образом на него.
Следует отметить, что осведомленность наших врачей об этом заболевании находится на достаточно высоком уровне. Во-первых, как выше сказано, ИТП описано 275 лет тому назад. Во-вторых, ИТП ‒ довольно распространенное среди редких заболевание. Ежегодно только в нашем отделении консультируются более 300 пациентов с диагнозом ИТП или с подозрением на нее. И наконец, впервые в России (в 2014 г.) разработаны Национальные клинические рекомендации (НКР) по диагностике и лечению идиопатической тромбоцитопенической пурпуры у взрослых наряду с другими нозологиями по инициативе Минздрава России.4 Данные рекомендации постоянно обновляются и общедоступны.4 Последняя редакция опубликована в 2021 году.
В 2021 году вышли обновленные клинические рекомендации Американской ассоциации гематологов (ASH) и обновленный Международный консенсус по диагностике и лечению первичной иммунной тромбоцитопении. Мы анализировали данные рекомендации и сравнивали с российскими клиническими рекомендациями по ИТП.
Хочу сказать, что глобальных изменений мы не заметили: особое внимание уделено правильной диагностике, а также более детально описаны подходы к лечению.
Кроме того, изучение ИТП входит в учебные и научные планы нашего Гематологического центра для гематологов, ординаторов, слушателей на циклах повышения квалификации ФГБУ «НМИЦ гематологии» Минздрава России и для специалистов других специальностей, поскольку имеются гематологические маски разных заболеваний. Наш Центр организует для врачей-специалистов стажировки на рабочем месте по актуальным вопросам гематологии как для гематологов, так и других специалистов. Все эти программы включены в систему непрерывного медицинского образования. В эру цифровизации и дистанционных образовательных технологий, проведения вебинаров, мастер-классов, обсуждения конкретных клинических примеров значительно расширяется аудитория обучающихся, что способствует непрерывному профессиональному росту врачей.
Лечение
Спленэктомия (СЭ), введенная в практику лечения еще в начале прошлого века Шлоффером, и сегодня является одним из вариантов лечения пациентов при ИТП. Она выполняется относительно часто, но в течение последних трех лет число таких вмешательств снизилась с 26 до 17%, при этом доля пациентов, получающих современные ЛС (агонисты тромбопоэтиновых рецепторов, аТПО) увеличилась с 5,9 до 45,7%. Такая же тенденция наблюдается и за рубежом, где частота выполнения спленэктомии меньше, чем в РФ.
С 1951 года в терапии заболевания используются кортикостероиды, до сих пор они остаются первой линией терапии пациентов с впервые диагностированной ИТП в зарубежных и российских протоколах – при геморрагическом синдроме и тромбоцитах менее 30‒50,0х109/л или при отсутствии геморрагического синдрома при тромбоцитопении 9/л.
Согласно данным нашего регистра, в 92,2% случаев в качестве первой линии пациенты получают лечение кортикостероидами как в виде стандартного лечения, так и пульс-терапии с эффективностью до 70‒80% с быстрым купированием геморрагического синдрома и повышением количества тромбоцитов выше безопасного уровня. Однако после отмены препарата быстро наступает рецидив заболевания. Кортикостероиды – эффективные средства, но с ними связано большое число потенциальных осложнений: сахарный диабет; тяжелые формы артериальной гипертензии и аритмий; язвенная болезнь ЖКТ, активные инфекции; психические расстройства. Поэтому повторные и частые курсы нежелательны. Во всех клинических рекомендациях строго ограничена продолжительность лечения кортикостероидами до 3‒4 недель. К сожалению, из-за доступности кортикостероиды назначаются и на последующих линиях терапии.
В 1980 году в университете при детской больнице в Берне 12-летнего мальчика с острой ИТП и иммунодефицитом лечили внутривенным иммуноглобулином (ВВИГ), что привело к заметному увеличению числа тромбоцитов в течение 24 часов. С тех пор ВВИГ широко и успешно применяется как в первой, так и в последующих линиях терапии в качестве «скорой помощи» в экстренных и жизнеугрожающих ситуациях и является совершенно неоценимым средством для лечения беременных с ИТП. ВВИГ в качестве первой линии терапии эффективен в 80% случаев, гемостатический эффект наступает на 1‒2-й день, длительность ответа – 1‒4 недели. Таким образом, фактически после первой линии терапии почти все пациенты – кандидаты второй линии терапии.
Для систематизации порядка назначения вариантов терапии международной рабочей группой по изучению ИТП выделены 3 стадии заболевания:
- впервые диагностированная с длительностью до 3 месяцев от момента диагностики;
- персистирующая с длительностью 3‒12 месяцев;
- хроническая с длительностью более 12 месяцев.
А последовательность назначения терапии при ИТП, разработанной на основании многолетнего клинического опыта, получила название линий терапии, которые в основном соответствуют стадиям заболевания.
В конце 2000 года, без сомнения, начинается новая эра лечения ИТП: препаратами современного поколения – стимуляторами тромбоцитопоэза, агонистами тромбопоэтиновых рецепторов, аТПО – ромиплостимом («Новартис») и элтромбопагом («Амджен»). В 2009 году они были одобрены в России как орфанные препараты для взрослых с рефрактерной хронической ИТП с/без спленэктомии. В 2015 году оба препарата включили в Перечень жизненно необходимых и важнейших лекарственных препаратов для медицинского применения, утверждаемый распоряжением Правительства Российской Федерации. Использование аТПО (эти данные основаны на доказательной медицине и подкреплены несколькими основательными и очень качественными проспективными контрольными исследованиями) эффективно как до спленэктомии, так и после нее. С появлением данных препаратов прогноз заболевания улучшился, так как они предотвращают развитие тяжелых побочных эффектов лечения и позволяют добиться 80%-ного уровня непосредственного эффекта.1,6
Я считаю (как и многие мои коллеги) их важными особенностями – органосохраняющий и кортикостероидсдерживающий эффекты.
В клинической практике сравнительно недавно появился еще один препарат для лечения ИТП – ритуксимаб, который разработан для лечения гематологических злокачественных заболеваний. В настоящее время ритуксимаб используется для лечения пациентов с ИТП, устойчивых к другим методам лечения. Его использование при хронической ИТП основывается на удалении аутореактивных В-лимфоцитов. Ритуксимаб включен в качестве 3-й линии терапии. Отмечается примерно 60%-ная вероятность получить первичный ответ. Но в России он не зарегистрирован для лечения ИТП, поэтому решение принимается индивидуально врачебной комиссией.
В 2021 году Управление по контролю качества пищевых продуктов и лекарственных средств США (FDA) одобрило новый пероральный лекарственный препарат – селективный низкомолекулярный ингибитор селезеночной тирозинкиназы, фостаматиниб – для медицинского применения у пациентов с резистентной ИТП. А в 2021 – биодоступный агонист малой молекулы рецептора тромбопоэтина, аватромбопаг – для лечения взрослых пациентов с хронической ИТП, которые имели недостаточный ответ на предыдущую терапию. Оба препарата в России не зарегистрированы для лечения ИТП. Это перспектива.
При неуспешности разных вариантов терапии в последующих линиях терапии рекомендуется использование нереализующего метода или проведение комплексной терапии с использованием иммуносупрессоров.
Как правило, современные методы терапии все же позволяют достигнуть ремиссии различной длительности или состояния клинической компенсации. Но четкие прогностические критерии течения заболевания, ответа на терапию и исходов болезни до сих пор не разработаны – из-за природы и непредсказуемого течения заболевания.
Приступая к терапии хронической, рецидивирующей ИТП, необходимо помнить, что выбор терапии должен быть направлен на купирование кровотечения любой локализации, улучшение качества жизни больного, а не на нормализацию количества тромбоцитов любой ценой.
В клинической практике важно помнить, что терапию всегда следует подбирать индивидуально для конкретного пациента с учетом его возраста, коморбидности, сопутствующей патологии, а также учитывать предпочтения больного. Но наша практика часто сталкивается с объективными реалиями жизни.
По данным Е.Ю. Красильниковой (руководителя проектного офиса «Редкие (орфанные) болезни» Национального НИИ общественного здоровья им. Н.А. Семашко), при подготовке Ежегодного бюллетеня по редким (орфанным) заболеваниям9 была получена информация от 76 регионов РФ, в которых на 1 января 2021 года проживало 3 860 пациентов с ИТП (из них 873 – дети), в лекарственной терапии нуждались 2069 человек (из них 468 – дети), получали лекарственную терапию 1 606 пациентов (из них 446 – дети). То есть патогенетическим лечением обеспечено 42% пациентов, внесенных в Федеральный регистр лиц, страдающих редкими жизнеугрожающими заболеваниями.8
Фактически более половины пациентов не получают предназначенное орфанное лечение. Качество оказания медицинской помощи, которое включает диагностику, определение тактики терапии, коррекцию и контроль над этими показателями, стало возможным благодаря разработке схемы маршрутизации пациента, которая представляет путь больного от установления диагноза до обеспечения необходимыми лекарственными препаратами, то есть от лечащего врача до включения пациентов в список федерального регистра.
Наш опыт работы с региональными гематологами показывает, что все они хорошо осведомлены и соблюдают пути маршрутизации пациентов для получения орфанных лекарственных препаратов. В трудных ситуациях они обращаются в федеральные центры для получения решения врачебной комиссии по дорогостоящим препаратам. Обеспечение же в регионах только 42% пациентов с ИТП современными дорогостоящими орфанными ЛС главным образом связано с недостаточным финансированием в ряде регионах. И это тоже имеет свое объяснение. Число пациентов, нуждающихся в дорогостоящих орфанных препаратах, благодаря улучшению их диагностики увеличивается. Единственный выход – включение ИТП в федеральную программу финансирования высокозатратных нозологий.
Таким образом, идиопатическая тромбоцитопеническая пурпура (ИТП) – редкое (орфанное) хроническое, рецидивирующее заболевание, значительно ухудшающее здоровье и качество жизни больных по оценкам физического, социального функционирования, психического состояния. Кровотечения вызывают у них страх, тревожность и депрессию при кратковременном эффекте от проведенной терапии и побочных эффектах лекарств на фоне длительного лечения кортикостероидами, иммуносупрессорами.
ИТП полностью излечить нельзя, но можно эффективно сдерживать. Появившиеся в последние годы современные лекарственные препараты (агонисты тромбопоэтиновых рецепторов) при адекватном выборе дозы и контроля течения заболевания позволяют быстро купировать геморрагический синдром, достигать ремиссии различной длительности или состояния клинической компенсации, предотвращать развитие тяжелых побочных эффектов лечения, улучшать прогноз заболевания, что, естественно, не только повышает продолжительность жизни пациентов с орфанными заболеваниями, но и ее качество. Поэтому очень важно их включение в терапию всех пациентов, нуждающихся в ней. Сегодня эта равная доступность возможна лишь при включении ИТП в федеральную программу финансирования высокозатратных нозологий.
Литература
- «Об основах охраны здоровья граждан в РФ» № 323-ФЗ от 21.11.2011. РГ, федеральный выпуск № 263 (5639) (от 23 ноября 2011 г.). https://rg.ru/2011/11/23/zdorovie-dok.html
- Постановление Правительства РФ № 403 от 26.04.2012 «О порядке ведения Федерального регистра лиц, страдающих жизнеугрожающими и хроническими прогрессирующими редкими (орфанными) заболеваниями, приводящими к сокращению продолжительности жизни граждан или их инвалидности, и его регионального сегмента». 2 мая 2012. https://www.garant.ru/products/ipo/prime/doc/70068888/
- Черников М.В., Куликов С.М., М. А. Русинов М.А. и соавт. Мультинозологический регистр заболеваний системы крови. Состав, структура, итоги опытной эксплуатации // Гематология и трансфузиология. Т. 59, № 1, 2014, с. 30.Меликян А.Л., Егорова Е.К., Пустовая Е.И., Колошейнова Т.И., Володичева Е.М., Капорская Т.С., Ильясов Р.К., Шелехова Т.В., Федорова Н.А., Зотова И.И., Сычева Т.М., Контиевский И.Н., Шестопалова И.А., Куркина Н.В., Сырцева Е.Б., Тарасенко Е.В. Промежуточные результаты эпидемиологического исследования идиопатической тромбоцитопенической пурпуры у взрослых в Российской Федерации // Гематология и трансфузиология. 2021. Т 64. № 4. С. 436‒446.
- Меликян А.Л., Пустовая Е.И., Егорова Е.К., Калинина М.В., Колошейнова Т.И., Суборцева И.Н., Гилязитдинова Е.А., Двирнык В.Н. Дифференциальная диагностика тромбоцитопений // Онкогематология. 2017;12(1):78‒87. https://doi.org/10.17650/1818-8346-2017-12-1-78-87
- Neunert C, Terrell DR, Arnold DM, et al. American Society of Hematology 2021 guidelines for immune thrombocytopenia. 2019;3(23):3829‒3866. doi:10.1182/bloodadvances.201900096
- Меликян А.Л., Пустовая Е.И., Егорова Е.К., Калинина М.В., Колошейнова Т.И., Суборцева И.Н., Гилязитдинова Е.А., Двирнык В.Н. Дифференциальная диагностика тромбоцитопений // Онкогематология. 2017;12(1):78‒87. https://doi.org/10.17650/1818-8346-2017-12-1-78-87
- Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3(22):3780‒3817. doi:10.1182/bloodadvances.2019000812
- Ежегодный бюллетень экспертного совета по редким (орфанным) заболеваниям. https://komitet2-.km.duma.gov.ru/upload/site21/Byulleten_po_redkim_zabolevaniyam_2020.pdf
- Клинические рекомендации «Идиопатическая тромбоцитопеническая пурпура (ИТП) у взрослых» (утв. Минздравом России), 2021. https://npngo.ru/uploads/media_document/283/5eb37419-9276-4e9a-b075-0e26a788f623.pdf
1.Общие сведения
Кровоточивое воспаление сосудистых стенок является достаточно частым заболеванием, – известным также под названиями болезнь Шенлейна-Геноха, пурпура Шенлейна-Геноха или анафилактический васкулит (в западной литературе и МКБ-10), геморрагический васкулит (в отечественной медицине), ревматическая или аллергическая пурпура, – которое манифестирует преимущественно в детском или раннем пубертатном возрасте.
Заболеваемость оценивается на уровне 0,2-0,25% среди лиц данной возрастной категории. Геморрагический васкулит считается одним из наиболее распространенных видов патологии сосудистых стенок. Поражаются мелкие сосуды кожи и внутренних органов; могут вовлекаться суставы, органы брюшной полости, почки. Заболевание впервые описано И.Л. Шёнляйном (1837) и затем более подробно изучено Е.Н.Генохом (1874), что и послужило основанием для наименования болезни.
Обязательно для ознакомления! Помощь в лечении и госпитализации!
Почему может появиться ревматическая пурпура?
Геморрагический васкулит, также именуется, как заболевание Шёнлейна-Геноха — разновидность воспалений мелких участков сосудистой системы.
Подобный процесс может возникнуть в теле человека вследствие сбоев в стабильной работе иммунной системы. К главным источникам развития болезни, относят:
- Острые и хронические болезни, вызванные бактериальной или вирусной инфекцией (например: ОРВИ, стафилококки, высыпания герпеса и прочее)$
- Наследственная предрасположенность;
- Отравление организма ядами биологического характера и химсредствами;
- Длительное нахождение на морозе или жаре;
- Укусы насекомых;
- Личностная реакция на ввод особых видов вакцинных препаратов;
- Солнечные ожоги кожного покрова.