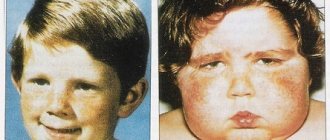
Синдром Ретта – это генетическое заболевание, сопровождающееся тяжелыми психоневрологическими симптомами. Диагностика его затруднительна: оно практически никогда не обнаруживается внутриутробно, а после рождения проявляется не ранее, чем через 6 месяцев. Своему носителю оно грозит глубоким слабоумием, двигательными ограничениями, дезадаптацией в социуме.
Истоки заболевания
В масштабном формате о расстройстве заговорили в 1983 году благодаря шведскому ученому Бенгту Хагбергу. В это время он со своей группой изучал 35 подобных между собой случаев в 3 разных странах: в Португалии, Франции и Швеции.
Однако Хагберт не является первооткрывателем синдрома. Впервые его обнаружил педиатр Андреас Ретт, имя которого носит заболевание. Он наблюдал за двумя девочками, имеющими одинаковые симптомы. Их он заметил в очереди на прием. Они сидели на коленях у матерей, а те держали их за руки. Девочки раскачивались как маятники, а затем внезапно обе начали совершать стереотипные движения руками. Дети застыли в одном положении, отстраненные от окружающего мира. Взгляд был направлен в одну точку. Поражала их синхронность в движениях и поведении.
В своих письменных архивах врач отыскал подобные истории болезни, а затем отправился в Европу, чтобы разыскать и там таких же пациентов. В 1966 он сделал первые публикации своих исследований, которые, однако, не вызвали особого интереса.
Зафиксированную им болезнь Ретт назвал синдромом атрофии мозга. Сначала ее считали проявлением аутизма или шизофрении, и только лишь в 1983 году вывели в отдельную нозологическую единицу.
В настоящее время синдром относят к категории довольно редких генетических заболеваний. Он встречается с частотой случаев 1 на 15000. Причиной его называют мутацию гена МЕСР2. Этот ген отвечает за синтез определенного белка, влияющего на развитие мозга. В норме этот белок, сп
устя некоторое время после рождения, должен подавляться другими генами, чтобы обеспечить нормальное развитие мозга.
Если же ген МЕСР2 мутирован, то белок инактивируется не полностью, что вызывает аномальное мозговое созревание, и провоцирует развитие синдрома Ретта.
Обычно мутирующий ген располагается в Х хромосоме, потому заболеванием страдают преимущественно девочки.
Помощь в
Задача родителей ребенка и специалистов центра — максимально сохранить физическое и психическое здоровье, не потерять контакт с ребенком. Для этого составляем индивидуальную программу терапии, учитывающую особенности ребенка.
В программу входят занятия в центре. В первую очередь — лечебная физкультура, остеопатическая коррекция и игротерапия. Занятия лечебной физкультурой и с остеопатом помогают сохранить двигательные функции, улучшить гибкость тела и амплитуду движения конечностей, поддержать мышечный тонус в норме. Игротерапия направлена на укрепление как физического, так и психоэмоционального состояния ребенка. Также проводим занятия с логопедом, дефектологом, клиническим психологом и нейропсихологом.
На каждом занятии обязательно участвуют родители. Помогаем им наладить контакт с ребенком, обучаем самостоятельной работе, чтобы необходимые упражнения выполнялись дома. Если у родителей возникли вопросы при выполнении домашних заданий, проконсультируем по телефону.
Почему мальчики не болеют
Учитывая, что мутирующий ген несет в себе Х-хромосома, то девочки в плане заболевания находятся в более «выигрышной» позиции. У них присутствует две Х-хромосомы. Поэтому если одна из них «бракованная», то вторая функционирует нормально. Это дает девочке хоть малый шанс на нормальное существование.
У мальчика Х-хромосома одна. Если она имеет мутационный ген, значит, выпадает из работы полностью, и ее нечем заменить. Такие малыши мужского пола, как правило, погибают еще внутриутробно, так и не родившись. Поэтому синдром Ретта у мальчиков встречается крайне редко.
Но, несмотря на такую особенность заболевания, очень редко, но все-таки мальчики с подобным синдромом выживают. Это может быть связано с тем, что не все гены в Х-хромосоме подвергаются мутации. Из-за этого заболевание развивается не столь остро.
Другая причина – наличие у мальчика синдрома Клайнфельтера. При этом наблюдается полисомия половых хромосом, то есть их набор составляет ХХУ. И, если одна Х-хромосома имеет патологический ген, то вторая может регулировать синтез белка и дарить мальчику возможность жизни. Получается такая же картина, как и у девочки.
Подходы к лечению и реабилитации при синдроме Ретта
Цели терапии:
- устранение психопатологических нарушений;
- достижение медикаментозной ремиссии;
- стабилизация состояния больного.
Тактика лечения при удовлетворительном и/или нетяжелом уровне синдрома Ретта основана на амбулаторной терапии. В случае усложненной клинической картины либо неэффективности амбулаторных мероприятий принимают решение о госпитализации. Лечение включает в себя медикаментозные методы и немедикаментозную терапию (комплаенс-терапия, психо-, трудотерапия).
В дальнейшем проводятся мероприятия по формированию и укреплению комплаенса (добровольного следования пациента предписанному режиму лечения). Требуется компетентный воспитательный подход, который помогает больным развить адаптивные навыки.
Как развивается заболевание
Синдром Ретта у детей – довольно коварное заболевание. При рождении оно практически не проявляет себя. Первые его симптомы появляются в период от 6 мес. до полутора лет. Однако некоторые, еле заметные признаки, в первом полугодии все-таки имеются. Но они настолько ничтожны, что не привлекают внимания.
Вот что говорит мама одной из девочек с синдромом по поводу первого полугодия ее жизни. Она придала значение этим мелочам только по прошествии 1 года и 7 месяцев с рождения ее дочери, когда проявления стали уже явными. Из предвестников болезни она отметила, что ее малышка начала держать голову в 3 месяца, а не в 2, как это положено. В 6 месяцев она еще не могла сидеть, а ходить начала только в 1 год и 4 месяца. Психологически развивалась нормально, и говорить начала рано, но это были не стандартные слова «мама», «папа», а «зайчик», «мишка» и др.
В 1 год и 7 мес. она перестала узнавать родителей и, казалось, не нуждалась в них. Весь день проводила за одним однообразным занятием: кидала мяч или катала коляску. Часами ходила по кругу, пока ее не останавливали или она запиналась. Такое стереотипное поведение носит название полевого, когда действие затягивает больного, и он не может ничего сделать.
В четыре года к симптомам присоединились эпилептоидные припадки. Однако по достижении школьного возраста девочка находилась на домашнем обучении, и делала некоторые успехи.
12–6 лет – это был период ремиссии, когда болезнь практически не беспокоила. Но с 16 лет появились новые, более глубокие проблемы, связанные с костными деформациями и болезнями внутренних органов. Одна нога девочки была короче другой почти на 10 см, что не могло не препятствовать ходьбе. В 20 лет она весила всего 24 кг с ростом 158 см.
Обычно СР протекает в 4 стадии.
Первая стадия, которая, как правило, стартует с 6 месяцев до полутора лет, проявляется нарастанием раздражительности и лабильностью настроения у ребенка. Эпизоды плача и психомоторного возбуждения сменяются все большей пассивностью. Малыш бесцельно передвигается по комнате, пропадает интерес к игрушкам. Но контакт с матерью сохраняется.
Вот как описывает женщина поведение своей дочери на заре заболевания: она кричала целый день без остановки, билась головой о стены, не могла уснуть. Что бы мы ни делали, она не успокаивалась. Это был настоящий ад. Но больше угнетало то, что ни один врач не мог поставить вразумительный диагноз.
Развивается диспропорция головы и конечностей по отношению к телу. Они становятся несоизмеримо маленькими. Замедляется рост, и снижается тонус мышц.
Вторая стадия, длящаяся несколько лет, отличается пестротой симптомов. Сразу обращает на себя внимание снижение интеллектуальных способностей, развивается умственное слабоумие. Происходит регресс практически всех полученных навыков. Речь полностью исчезает или переходит в степень эхолалии – механического повторения услышанного.
Приобретенные двигательные навыки, предметно-ролевое поведение теряются и замещаются двигательными стереотипами. Характерный симптом: многочисленно повторяющиеся движения, напоминающие мытье рук. Кроме этого, ребенок постоянно заламывает или потирает их, размахивает ими, хлопает в ладоши. Сжатие пальцев рук вполне нормально в 4 месяца, но в более позднем возрасте говорит об остановке развития. Малыш утрачивает хватательный рефлекс, не способен производить вращательные движения руками.
Постепенно двигательная активность сходит на нет. Нарушается походка, ребенок ходит, не сгибая коленей.
Третья стадия длится 10 лет и более, характеризуется она развитием стойкого, глубокого слабоумия, вплоть до идиотии. Наблюдается полная потеря способности говорить и понимать обращенную к ребенку речь. Появляется тремор всего тела, отягчающий движения. Усиливаются судорожные припадки.
Четвертая, конечная стадия – это период усугубления ранее проявляемых симптомов. Стойкая утрата умственных способностей, двигательных навыков, развитие мышечных дистрофий, приводящих к полному обездвиживанию.
Продолжительность жизни таких больных в среднем колеблется до 30 лет, хотя известны случаи, когда они доживали и до 50-летнего возраста.
Первичный иммунодефицит
Первичные иммунодефициты — гетерогенная группа редких заболеваний, преимущественно связанных с врожденными генетическими дефектами иммунной системы. Клинически ПИД могут проявляться иммунологическими нарушениями, пороками развития, гематологическими и аутоиммунными заболеваниями, атопиями, злокачественными новообразованиями. Чаще всего ПИД выявляют еще в младенческом возрасте, основными признаками являются частые и рецидивирующие инфекции с тяжелым течением, задержка роста и развития, желудочно-кишечные расстройства.
Подробнее о причинах, симптомах, диагностике и лечении первичных иммунодефицитов можно прочитать здесь
Первичный иммунодефицит
Самые частые симптомы расстройства
Типичные симптомы для синдрома Ретта – мышечные и двигательные нарушения. Мышцы находятся в гипертонусе или же, наоборот, теряют его. В этом случае у ребенка развивается неправильное положение тела, прогрессирует частичные параличи и нарушение координации. Например, девочки скрещивают ноги во время ходьбы.
Синкинезии – патологические сокращения мышц, возникают вслед за произвольным движением: простая улыбка способна вызвать резкий взмах ногой. Такое явление постепенно приводит к повреждению суставов, сухожилий и связок, провоцирует ортопедические нарушения. Последние проявляются во всевозможных деформациях и также очень часто сопровождают таких детей. Среди них выделяют вывих тазобедренного сустава, провоцируемый малой подвижностью.
Статическая деформация стопы чаще развивается из-за нарушенного мышечного тонуса. Распространенной считается патология под названием «конская стопа», связанная со снижением подвижности голеностопного сустава. Ее можно узнать по пятке, которая не достигает земли, стопа при этом смещается кнаружи или вовнутрь. Причина патологии – гипертонус икроножной мышцы.
Сколиоз – боковое искривление позвоночника, который провоцирует массу проблем у таких пациентов: деформации суставов и костей, боли во время ходьбы, в стоячем или сидячем положении, утрата способности передвигаться. Сколиоз грудного отдела вызывает легочную недостаточность. Появляются также проблемы с пищеварением.
У детей с синдромом Ретта наблюдается повышенное слюнотечение. Но это происходит не из-за избытка количества слюны, а потери способности сглатывать ее.
Нарушение питания может развиваться из-за частых приступов тошноты. Она появляется на любые аспекты питания: на определенный продукт, его температуру, на способ приготовления. Так, ребенок способен отрицательно реагировать на пищу, поданную кусочками, или на комочки в блюде.
Постоянная тошнота провоцирует отказ от питания, а значит, потерю в весе.
Плохое сглатывание слюны, которая регулирует кислотность в желудке, и повышенное внутрибрюшное давление вызывают желудочно-пищеводный рефлюкс, то есть забрасывание содержимого желудка в пищевод. Это чревато такими последствиями, как воспаление стенки пищевода, респираторные инфекции.
Малоподвижный образ жизни, неврологические расстройства, неправильное питание провоцируют возникновение запоров у детей с синдромом Ретта. Они носят тяжелый характер, поскольку способны вызывать закупорку кишечника и сильные боли.
Повышенное слюнотечение, тошнота, рефлюкс снижают потребление ребенком пищи и даже развивают на нее негативную реакцию. В результате этого ребенок теряет в весе. Этот процесс стоит строго контролировать, поскольку он чреват истощением.
Другое тяжелое расстройство связано с работой дыхательной системы, развивающееся вплоть до приступов апноэ. Это явление настолько часто среди детей с синдромом, что нередко стает причиной их гибели.
Важными патогномоничными признаками синдрома считаются проявления аутизма. Именно из-за них заболевание изначально считали одной из форм этого расстройства, а в настоящее время относят к болезням аутистического спектра.
Аутистические признаки проявляются в отстранении от окружающего мира, в том числе и от родственников. Ребенок замыкается в себе, может не откликаться, когда его зовут. Предпочитает одиночество. Дети боятся чужих людей и непривычных ситуаций.
Лицо такого ребенка становится похожим на каменное. Взгляд блуждающий или устремлен в одну точку. Поведение часто непредсказуемо: случаются приступы неутомимого смеха или плача. Склонны к самоповреждениям: царапают кожу, кусают пальцы, вырывают волосы.
Интерстициальные болезни легких у детей
Интерстициальные болезни легких у детей — гетерогенная (неоднородная) группа редких заболеваний легких у младенцев, детей и подростков. Их развитие приводит к нарушениям газообмена и вентиляционной функции легких, диффузным изменениям интерстициальной ткани легких.
К ИБЛ, чаще встречающимся в младенческом возрасте, относятся врожденные нарушения функции сурфактанта, связанные с мутациями, ацинарная дисплазия, врожденная альвеолярная дисплазия, альвеолярно-капиллярная дисплазия с нарушением месторасположения легочных вен, аномалии альвеолярного роста, нейроэндокринная гиперплазия, легочный интерстициальный гликогенез.
У детей старше 2 лет и подростков встречаются следующие ИБЛ: идиопатические интерстициальные пневмонии (неспецифическая интерстициальная пневмония, десквамативная интерстициальная пневмония, криптогенная организующаяся пневмония, лимфоцитарная интерстициальная пневмония); синдромы альвеолярного кровотечения, синдромы аспирации, гиперчувствительный пневмонит, фолликулярный бронхиолит, эозинофильная пневмония, альвеолярный протеиноз, лимфангиолейомиоматоз, лимфангиэктазия, гемангиоматоз; патологии, связанные с системными заболеваниями (саркоидоз легких, поражения легких при склеродермии, гистиоцитоз легких, заболевания легких, вызванные злокачественными новообразованиями); иммуноопосредованные заболевания легких; заболевания легких, связанные с ослаблением иммунной системы.
Причины, по которым могут развиваться интерстициальные болезни легких у детей, во многих случаях неизвестны. К условиям или факторам, которые могут влиять на возникновение ИБЛ, относятся унаследованные состояния (врожденные нарушения функции сурфактанта), врожденные состояния, вызывающие нарушения структуры и функции легких, синдромы аспирации, различные нарушения работы иммунной системы, неблагоприятное воздействие окружающей среды, некоторые методы лечения рака (химиотерапия, лучевая терапия), системные или аутоиммунные заболевания, семейный анамнез ИБЛ.
Поскольку ИБЛ у детей имеют разнообразные формы, признаки и симптомы могут различаться. К основным симптомам относятся: учащенное поверхностное дыхание (тахипноэ), дыхательная недостаточность (респираторный дистресс-синдром), низкий уровень содержания кислорода в крови (гипоксемия), периодический кашель, хрипы с крепитацией (потрескиванием, хрустом, хорошо различимыми при выслушивании легких), одышка при физической нагрузке (во время еды у младенцев), замедленный рост, неспособность набрать вес, рецидивирующие пневмония или бронхиолит.
Диагноз ИБЛ у детей устанавливается на основании истории болезни, семейного анамнеза, результатов лабораторных (в т. ч. генетических) и инструментальных исследований (рентгенография органов грудной клетки, КТ), функциональных тестов легких, важным критерием постановки диагноза является исключение других заболеваний со схожей симптоматикой. Если диагностика не дает полной информации, врач может порекомендовать проведение биопсии легкого с последующим гистологическим исследованием.
Лечением интерстициальных болезней легких у детей занимается врач-пульмонолог.
Поскольку ИБЛ у детей относятся к редким заболеваниям, за недостаточностью исследований как отдельных форм заболевания, так и группы в целом, действенной терапии не существует. Современный подход в лечении основан на применении симптоматической и поддерживающей терапии, помогающих уменьшить проявление симптомов и повысить качество жизни пациента, максимально улучшить условия для нормального роста и развития, не допустить развития предотвратимых заболеваний, которые могут серьезно ухудшить состояние ребенка.
Поддерживающее лечение включает кислородную терапию; лекарства (бронходилататоры — препараты, расслабляющие гладкую мускулатуру бронхов, расширяющие их просвет и устраняющие бронхоспазм); налаживание питания, способствующее набору веса и нормальному росту ребенка; аппараты для отведения мокроты; аппараты НИВЛ и ИВЛ; легочную реабилитацию.
Симптоматическое лечение включает кортикостероиды, помогающие уменьшить воспаление в легких; антибиотики для лечения инфекции; препараты для предотвращения кислотного рефлюкса, опасного аспирацией, и другие. Трансплантация легких — на сегодня единственный эффективный метод лечения пациентов с такими жизнеугрожающими состояниями, как дисплазия альвеолярных капилляров и нарушения функции сурфактанта, вызванные определенными мутациями генов.
Важно принять меры профилактики заболеваний (в первую очередь инфекционных), которые могут ухудшить состояние ребенка: мыть руки; держаться подальше от людей с симптомами простуды; обсудить с педиатром план вакцинации не только ребенка, но и окружающих его близких; поговорить с врачом о том, как обезопасить ребенка от респираторно-синцитиальной (вирусной) инфекции, которая у здоровых людей может вызвать симптомы простуды и гриппа, а у пациента с ИБЛ серьезно ухудшить состояние; избегать воздействия загрязненного воздуха, табачного дыма, других веществ, раздражающих легкие.
«Когда в Европе был локдаун, мы делились друг с другом лекарственными запасами»
— Чем лечат детей с мутацией в гене CDKL5?
— От мутации лекарства пока нет, но нашим детям необходимы препараты от эпилепсии. И большинство этих препаратов — не зарегистрированные в России. Нам приходится их доставать, часто подпольно. Помните, одна мать заказала из-за границы фризиум, и на нее дело завели? Он же тоже против эпилепсии. Вот наши родители каждый раз рискуют. Есть федеральный фонд «Круг добра», в который мы обратились с просьбой помочь нам с лекарствами, но они нас не приняли в фонд, потому что у нас мутация неизлечимая, а они принимают только тех, у кого заболевание лечится. Наша мутация — жизнеугрожающее, редкое, неизлечимое заболевание. Но и в нашем царстве слез есть луч надежды — это международный фонд LouLou, основанный семьей Джафар, в которой тоже родился ребенок с мутацией CDKL5. Это очень богатая ливанская семья — муж, Мажид Джафар, из Ирака, жена, Линн, — и они оплачивают все мировые исследования, в рамках которых ученые пытаются найти препарат против этой мутации, а также разрабатывают новые противоэпилептические препараты. 18 лабораторий мира работают за счет грантов этого фонда. Мы тоже с ними знакомы, они проводят международные научные форумы, ездят на них, оплачивают для семей, которые хотят попасть на эти форумы, проезд и проживание. Мы от нашей Ассоциации ездим туда бесплатно, они оплачивают билеты для главы ассоциации, врача, а, когда мы летали в Бостон, оплатили еще участие нашего переводчика и двух волонтеров. Мы получаем на этих форумах самую последнюю информацию, переводим ее и размещаем в группе, так что наши родители знают, какие исследования идут, на каком они этапе и так далее.
— И на каком этапе находятся эти исследования?
— Сейчас несколько лабораторий разработали препараты против этой мутации, один уже испытывают на животных.
Наши ученые-генетики тоже хотят участвовать в таких исследованиях, у них большой интерес, у нас мощная генетика, мировое сообщество готово давать гранты на такие исследования в России, но почему-то в нашей стране такие исследования не приветствуются.
Часто нам говорят, что у государственных учреждений нет валютного счета, и они просто не могут такие гранты принять.
— А на каком уровне вы сталкиваетесь с такими опасениями? На федеральном?
— Нет, скорее на уровне Московского департамента здравоохранения, они почему-то боятся делать совместные исследования, не хотят, чтобы наши генетики получали гранты, например, от американцев. Хотя Пенсильванский университет и фонд LouLou дают ежегодные гранты на такие исследования. А ведь было бы здорово работать над этим вместе.
— Теми препаратами, которые уже создаются, можно будет вылечить детей с мутацией гена CDKL5?
Как корреспондент “Ъ” провела день с подопечным детского хосписа
— Есть препарат ганаксалон — его исследовали в НИКИ педиатрии, он показал снижение количества приступов на 30%. И мы ждем, что в этом году он уже появится на российском рынке. Пока неизвестно, сколько он будет стоить, как его будут выписывать. Я надеюсь, что процесс закупки лекарств, выдачи льготных рецептов будет не сложным для родителей. Сейчас выбить какой-то рецепт — это целый подвиг. Я уже не говорю про отношение: ты приходишь в поликлинику, в соцзащиту — и как будто сразу всем там что-то должен. Ни о каком дружелюбии речи не идет. А хотелось бы прийти туда, где тебе помогут, где тебя защитят и подскажут, как правильно действовать.
— Вы сказали, что у вас в ассоциации 68 семей, воспитывающих детей с мутацией CDKL5. А в России их сколько?
— Тысячи. 68 — это число выявленных, диагностированных детей, живущих в крупных городах, которые нашли друг друга. В мире их — миллионы. Если один случай на 40 тысяч, посчитайте, сколько их может быть в России. И число детей с таким диагнозом растет по всему миру.
— Какой самый взрослый ребенок с такой мутацией живет сейчас в России?
— У нас в ассоциации такому ребенку 16 лет. А самому маленькому — полгода. Многие семьи, кстати, рожают других, здоровых, детей. Но женщины с трудом на это решаются, им нужен психолог. Мы сами в своем сообществе поддерживаем этих женщин, они испытывают огромный страх.
— А пренатальный скрининг не делают?
— Делают, и на эту мутацию нам тоже предлагают сделать. Но не все готовы на такое вмешательство, это же травматичная процедура.
«Департаменты соцзащиты работают на то, чтобы никого не защищать»
— Как вам кажется, в обществе отношение к детям и взрослым с инвалидностью меняется?
— В прошлом году мы провели первую встречу семей, воспитывающих детей с мутацией CDKL5 в России. Семьи из 28 городов приехали в Москву, с билетами и гостиницей нам помог как раз фонд LouLou. Многие приехали с другими детьми, с отцами. Мне очень понравилось, что у многих полные семьи, отцы их не бросают, помогают и дают своим женам отдохнуть. И в этом году, 1 июня, мы готовим вторую такую встречу, на нее приедет 145 человек.
Но было очень сложно найти отель для нашего мероприятия. Единственный отель, который согласился принять наши семьи,— Holiday Inn на Павелецкой. Мы отправили порядка 50 писем в разные отели, но нам отвечали: «У нас нет пандусов», «Мы переполнены». На самом деле причина, конечно, не в этом. Просто общество пока не готово видеть детей в тяжелом состоянии, в инвалидных колясках.