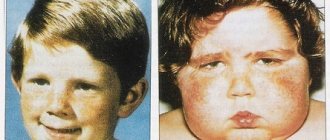
Rett syndrome is a genetic disease accompanied by severe neuropsychiatric symptoms. Diagnosing it is difficult: it is almost never detected in utero, and after birth it appears no earlier than 6 months later. It threatens its bearer with profound dementia, motor limitations, and maladjustment in society.
Origins of the disease
They started talking about the disorder on a large scale in 1983 thanks to the Swedish scientist Bengt Hagberg. At this time, he and his group studied 35 similar cases in 3 different countries: Portugal, France and Sweden.
However, Hagbert is not the discoverer of the syndrome. It was first discovered by pediatrician Andreas Rhett, whose name the disease bears. He observed two girls who had the same symptoms. He noticed them in the reception line. They sat on their mothers' laps, and they held their hands. The girls swung like pendulums, and then suddenly both began making stereotypical movements with their hands. The children were frozen in one position, detached from the world around them. The gaze was directed to one point. Their synchronicity in movements and behavior was amazing.
The doctor found similar case histories in his written archives, and then went to Europe to find the same patients there. In 1966, he made the first publications of his research, which, however, did not arouse much interest.
Rett called the disease he recorded brain atrophy syndrome. At first it was considered a manifestation of autism or schizophrenia, and only in 1983 was it brought into a separate nosological unit.
Currently, the syndrome is classified as a rather rare genetic disease. It occurs with a incidence of 1 in 15,000. The cause is said to be a mutation in the MECP2 gene. This gene is responsible for the synthesis of a certain protein that affects brain development. Normally this protein is
established some time after birth, must be suppressed by other genes to ensure normal brain development.
If the MECP2 gene is mutated, the protein is not completely inactivated, which causes abnormal brain maturation and provokes the development of Rett syndrome.
Usually the mutating gene is located on the X chromosome, so the disease mainly affects girls.
Help in
The task of the child’s parents and the center’s specialists is to preserve physical and mental health as much as possible and not lose contact with the child. To do this, we draw up an individual therapy program that takes into account the child’s characteristics.
The program includes classes at the center. First of all, physical therapy, osteopathic correction and play therapy. Exercise therapy and with an osteopath help maintain motor functions, improve body flexibility and range of motion of the limbs, and maintain normal muscle tone. Game therapy is aimed at strengthening both the physical and psycho-emotional state of the child. We also conduct classes with a speech therapist, speech pathologist, clinical psychologist and neuropsychologist.
Parents must participate in every lesson. We help them establish contact with the child, teach them how to work independently so that the necessary exercises can be done at home. If parents have any questions about doing homework, we will advise you by phone.
Why don't boys get sick?
Considering that the mutating gene is carried on the X chromosome, girls are in a more “advantageous” position in terms of the disease. They have two X chromosomes. Therefore, if one of them is “defective,” then the second one functions normally. This gives the girl at least a small chance for a normal life.
The boy has one X chromosome. If it has a mutation gene, it means it is completely out of work, and there is nothing to replace it. Such male babies, as a rule, die in utero, without being born. Therefore, Rett syndrome is extremely rare in boys.
But, despite this peculiarity of the disease, it is very rare that boys with a similar syndrome survive. This may be due to the fact that not all genes on the X chromosome undergo mutation. Because of this, the disease does not develop as acutely.
Another reason is that the boy has Klinefelter syndrome. In this case, polysomy of sex chromosomes is observed, that is, their set is XXY. And, if one X chromosome has a pathological gene, then the second can regulate protein synthesis and give the boy the opportunity to live. The result is the same picture as the girl's.
Approaches to treatment and rehabilitation for Rett syndrome
Goals of therapy:
- elimination of psychopathological disorders;
- achieving drug remission;
- stabilization of the patient's condition.
Treatment tactics for satisfactory and/or non-severe levels of Rett syndrome are based on outpatient therapy. In case of a complicated clinical picture or ineffectiveness of outpatient measures, a decision is made on hospitalization. Treatment includes drug methods and non-drug therapy (compliance therapy, psychotherapy, occupational therapy).
In the future, measures are taken to form and strengthen compliance (the patient’s voluntary adherence to the prescribed treatment regimen). A competent educational approach is required that helps patients develop adaptive skills.
How the disease develops
Rett syndrome in children is a rather insidious disease. At birth, it practically does not manifest itself. Its first symptoms appear within 6 months. up to one and a half years. However, there are still some barely noticeable signs in the first half of the year. But they are so insignificant that they do not attract attention.
This is what the mother of one of the girls with the syndrome says about the first half of her life. She attached importance to these little things only after 1 year and 7 months had passed since the birth of her daughter, when the manifestations had already become obvious. Among the harbingers of the disease, she noted that her baby began to hold her head at 3 months, and not at 2, as expected. At 6 months she could not sit yet, and she began to walk only at 1 year and 4 months. She developed normally psychologically, and began to speak early, but these were not the standard words “mom”, “dad”, but “bunny”, “bear”, etc.
At 1 year and 7 months. she stopped recognizing her parents and did not seem to need them. I spent the whole day doing one monotonous activity: throwing a ball or pushing a stroller. She walked in circles for hours until she was stopped or she stammered. This stereotypical behavior is called field behavior, when the action drags on the patient and he cannot do anything.
At the age of four, epileptoid seizures joined the symptoms. However, upon reaching school age, the girl was home-schooled and made some progress.
12–6 years – this was a period of remission, when the disease practically did not bother me. But from the age of 16, new, deeper problems appeared related to bone deformations and diseases of the internal organs. One of the girl’s legs was almost 10 cm shorter than the other, which could not but interfere with walking. At 20 years old, she weighed only 24 kg and was 158 cm tall.
Typically, SR occurs in 4 stages.
The first stage , which, as a rule, starts from 6 months to one and a half years, is manifested by an increase in irritability and lability of mood in the child. Episodes of crying and psychomotor agitation are replaced by increasing passivity. The baby moves aimlessly around the room and loses interest in toys. But contact with the mother remains.
This is how a woman describes her daughter’s behavior at the beginning of the disease: she screamed all day long without stopping, banged her head against the walls, and could not sleep. No matter what we did, she did not calm down. It was real hell. But what was more depressing was that not a single doctor could make a clear diagnosis.
A disproportion of the head and limbs in relation to the body develops. They become disproportionately small. Growth slows and muscle tone decreases.
The second stage , which lasts several years, is characterized by a variety of symptoms. A decrease in intellectual abilities immediately attracts attention, and mental dementia develops. Almost all acquired skills regress. Speech completely disappears or progresses to the level of echolalia - mechanical repetition of what was heard.
Acquired motor skills and object-role behavior are lost and replaced by motor stereotypes. A characteristic symptom: numerous repetitive movements reminiscent of hand washing. In addition, the child constantly wrings or rubs them, waves them, and claps his hands. Clenching of the fingers is quite normal at 4 months, but at a later age it indicates a stoppage of development. The baby loses his grasp reflex and is unable to make rotational movements with his hands.
Gradually, physical activity fades away. The gait is disturbed, the child walks without bending his knees.
The third stage lasts 10 years or more, and is characterized by the development of persistent, profound dementia, even idiocy. There is a complete loss of the ability to speak and understand speech addressed to the child. A tremor of the whole body appears, aggravating movements. Convulsive seizures intensify.
The fourth, final stage is a period of worsening of previously manifested symptoms. Persistent loss of mental abilities, motor skills, development of muscular dystrophies leading to complete immobility.
The life expectancy of such patients on average ranges up to 30 years, although there are cases when they lived up to 50 years of age.
Primary immunodeficiency
Primary immunodeficiencies are a heterogeneous group of rare diseases primarily associated with congenital genetic defects of the immune system. Clinically, PID can manifest as immunological disorders, developmental defects, hematological and autoimmune diseases, atopies, and malignant neoplasms. Most often, PID is detected in infancy; the main signs are frequent and recurrent infections with a severe course, delayed growth and development, and gastrointestinal disorders.
You can read more about the causes, symptoms, diagnosis and treatment of primary immunodeficiencies here
Primary immunodeficiency
The most common symptoms of the disorder
Typical symptoms of Rett syndrome are muscle and movement disorders. The muscles are in hypertonicity or, conversely, lose it. In this case, the child develops abnormal body position, partial paralysis and impaired coordination progress. For example, girls cross their legs while walking.
Synkinesias are pathological muscle contractions that occur following voluntary movement: a simple smile can cause a sharp swing of the leg. This phenomenon gradually leads to damage to joints, tendons and ligaments, and provokes orthopedic disorders. The latter manifest themselves in all sorts of deformations and also very often accompany such children. Among them are dislocation of the hip joint, provoked by low mobility.
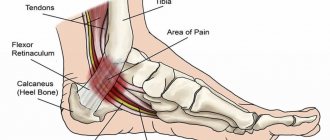
Static foot deformity often develops due to impaired muscle tone. A common pathology called “cauda foot” is considered to be associated with decreased mobility of the ankle joint. It can be recognized by the heel, which does not reach the ground, while the foot moves outward or inward. The cause of the pathology is hypertonicity of the calf muscle.
Scoliosis is a lateral curvature of the spine, which provokes a lot of problems in such patients: deformation of joints and bones, pain while walking, in a standing or sitting position, loss of the ability to move. Thoracic scoliosis causes pulmonary failure. Digestive problems also appear.
Children with Rett syndrome experience increased drooling. But this is not due to an excess amount of saliva, but a loss of the ability to swallow it.
Eating disorders can develop due to frequent attacks of nausea. It appears on any aspect of nutrition: on a certain product, its temperature, on the method of preparation. Thus, a child is able to react negatively to food served in pieces or to lumps in a dish.
Constant nausea provokes a refusal to eat, which means weight loss.
Poor swallowing of saliva, which regulates acidity in the stomach, and increased intra-abdominal pressure cause gastroesophageal reflux, that is, the reflux of stomach contents into the esophagus. This is fraught with consequences such as inflammation of the esophageal wall and respiratory infections.
A sedentary lifestyle, neurological disorders, and poor nutrition provoke constipation in children with Rett syndrome. They are severe because they can cause intestinal blockage and severe pain.
Increased salivation, nausea, and reflux reduce the child’s food intake and even develop a negative reaction to it. As a result, the child loses weight. This process should be strictly controlled, as it is fraught with exhaustion.
Another severe disorder is associated with the functioning of the respiratory system, developing up to attacks of apnea. This phenomenon is so common among children with the syndrome that it often causes their death.
Manifestations of autism are considered important pathognomonic signs of the syndrome. It is because of them that the disease was initially considered one of the forms of this disorder, and is now classified as an autism spectrum disorder.
Autistic signs manifest themselves in withdrawal from the outside world, including from relatives. The child withdraws into himself and may not respond when his name is called. Prefers solitude. Children are afraid of strangers and unusual situations.
The face of such a child becomes like stone. The gaze is wandering or fixed on one point. Behavior is often unpredictable: there are bouts of tireless laughter or crying. Prone to self-harm: scratching skin, biting fingers, pulling out hair.
Interstitial lung diseases in children
Interstitial lung diseases in children are a heterogeneous (heterogeneous) group of rare lung diseases in infants, children and adolescents. Their development leads to disturbances in gas exchange and ventilation function of the lungs, diffuse changes in the interstitial tissue of the lungs.
IPDs that are more common in infancy include congenital surfactant dysfunction associated with mutations, acinar dysplasia, congenital alveolar dysplasia, alveolar-capillary dysplasia with a violation of the location of the pulmonary veins, alveolar growth abnormalities, neuroendocrine hyperplasia, pulmonary interstitial glycogenesis.
The following IPDs occur in children over 2 years of age and adolescents: idiopathic interstitial pneumonia (nonspecific interstitial pneumonia, desquamative interstitial pneumonia, cryptogenic organizing pneumonia, lymphocytic interstitial pneumonia); alveolar hemorrhage syndromes, aspiration syndromes, hypersensitivity pneumonitis, follicular bronchiolitis, eosinophilic pneumonia, alveolar proteinosis, lymphangioleiomyomatosis, lymphangiectasia, hemangiomatosis; pathologies associated with systemic diseases (pulmonary sarcoidosis, lung lesions due to scleroderma, pulmonary histiocytosis, lung diseases caused by malignant neoplasms); immune-mediated lung diseases; lung diseases associated with a weakened immune system.
The reasons why interstitial lung disease may develop in children are in many cases unknown. Conditions or factors that may influence the occurrence of IPD include inherited conditions (congenital surfactant disorders), congenital conditions that cause changes in lung structure and function, aspiration syndromes, various immune system disorders, environmental exposures, and some cancer treatments. (chemotherapy, radiation therapy), systemic or autoimmune diseases, family history of IPD.
Because IPD comes in many forms in children, signs and symptoms may vary. The main symptoms include: rapid shallow breathing (tachypnea), respiratory failure (respiratory distress syndrome), low levels of oxygen in the blood (hypoxemia), intermittent cough, wheezing with crepitus (crackling, crunching, clearly distinguishable when listening to the lungs), shortness of breath during exercise (while eating in infants), slow growth, inability to gain weight, recurrent pneumonia or bronchiolitis.
The diagnosis of IPD in children is established on the basis of medical history, family history, results of laboratory (including genetic) and instrumental studies (chest x-ray, CT), pulmonary function tests; an important criterion for making a diagnosis is the exclusion of other diseases with similar symptoms . If the diagnosis does not provide complete information, the doctor may recommend a lung biopsy followed by histological examination.
A pulmonologist treats interstitial lung diseases in children.
Since IPD in children is a rare disease, due to insufficient research on both individual forms of the disease and the group as a whole, there is no effective therapy. The modern approach to treatment is based on the use of symptomatic and supportive therapy to help reduce symptoms and improve the patient’s quality of life, maximize conditions for normal growth and development, and prevent the development of preventable diseases that can seriously worsen the child’s condition.
Supportive treatment includes oxygen therapy; medications (bronchodilators - drugs that relax the smooth muscles of the bronchi, expand their lumen and eliminate bronchospasm); establishing nutrition that promotes weight gain and normal growth of the child; sputum removal devices; NIV and ventilator devices; pulmonary rehabilitation.
Symptomatic treatment includes corticosteroids, which help reduce inflammation in the lungs; antibiotics to treat infection; drugs to prevent acid reflux, which is dangerous due to aspiration, and others. Lung transplantation is currently the only effective treatment for patients with life-threatening conditions such as alveolar capillary dysplasia and surfactant dysfunction caused by certain gene mutations.
It is important to take measures to prevent diseases (primarily infectious) that can worsen the child’s condition: wash your hands; stay away from people with cold symptoms; Discuss with your pediatrician a vaccination plan not only for the child, but also for those around him; talk to your doctor about how to protect your child from respiratory syncytial (viral) infection, which can cause cold and flu symptoms in healthy people, but can seriously worsen the condition in a patient with IPD; Avoid exposure to polluted air, tobacco smoke, and other substances that irritate the lungs.
“When there was a lockdown in Europe, we shared medicine supplies with each other”
— How are children with a mutation in the CDKL5 gene treated?
“There is no cure for the mutation yet, but our children need drugs for epilepsy. And most of these drugs are not registered in Russia. We have to get them, often underground. Remember, one mother ordered freesium from abroad, and a case was opened against her? He is also against epilepsy. Our parents take risks every time. There is a federal fund “Circle of Good”, which we asked to help us with medicines, but they did not accept us into the fund because we have an incurable mutation, and they only accept those whose disease is being treated. Our mutation is a life-threatening, rare, incurable disease. But in our kingdom of tears there is a ray of hope - this is the international LouLou foundation, founded by the Jafar family, who also had a child with the CDKL5 mutation. This is a very rich Lebanese family - the husband, Majid Jafar, is from Iraq, the wife, Lynn - and they are paying for all the world's research in which scientists are trying to find a drug against this mutation, and are also developing new anti-epileptic drugs. 18 laboratories around the world operate with grants from this fund. We are also familiar with them, they hold international scientific forums, travel to them, and pay for travel and accommodation for families who want to attend these forums. We from our Association go there for free, they pay for tickets for the head of the association, the doctor, and when we flew to Boston, they also paid for the participation of our translator and two volunteers. We get the latest information from these forums, translate it and post it in the group, so our parents know what research is going on, what stage it is at, and so on.
— And at what stage are these studies?
“Several laboratories have now developed drugs against this mutation, one is already being tested on animals.
Our genetic scientists also want to participate in such research, they have great interest, we have powerful genetics, the world community is ready to give grants for such research in Russia, but for some reason such research is not welcomed in our country.
We are often told that government agencies do not have a foreign currency account and they simply cannot accept such grants.
— At what level do you encounter such concerns? At the federal one?
- No, rather at the level of the Moscow Department of Health, for some reason they are afraid to do joint research, they do not want our geneticists to receive grants, for example, from the Americans. Although the University of Pennsylvania and the LouLou Foundation provide annual grants for such research. But it would be great to work on this together.
— With the drugs that are already being created, will it be possible to cure children with a mutation of the CDKL5 gene?
How a Kommersant correspondent spent the day with a child’s hospice ward
— There is a drug ganaxalone - it was studied at the Pediatrics Research Institute, it showed a reduction in the number of attacks by 30%. And we expect it to appear on the Russian market this year. It is not yet known how much it will cost, how it will be prescribed. I hope that the process of purchasing medicines and issuing preferential prescriptions will not be difficult for parents. Nowadays, knocking out a recipe is quite a feat. I'm not even talking about the attitude: you come to the clinic, to the social security office - and it’s as if you immediately owe something to everyone there. There is no talk of any friendliness. But I would like to come to a place where they will help you, where they will protect you and tell you how to act correctly.
— You said that in your association there are 68 families raising children with the CDKL5 mutation. How many are there in Russia?
- Thousands. 68 is the number of identified, diagnosed children living in large cities who have found each other. There are millions of them in the world. If there is one case in 40 thousand, count how many there could be in Russia. And the number of children with this diagnosis is growing around the world.
— What is the oldest child with such a mutation currently living in Russia?
— In our association, such a child is 16 years old. And the youngest is six months old. Many families, by the way, give birth to other, healthy children. But women have a hard time deciding to do this; they need a psychologist. We ourselves in our community support these women, they experience great fear.
— Don’t they do prenatal screening?
- They do, and they also offer us to do this mutation. But not everyone is ready for such an intervention; it is a traumatic procedure.
“Social Security Departments are working to protect no one.”
— Do you think society’s attitude towards children and adults with disabilities is changing?
— Last year we held the first meeting of families raising children with the CDKL5 mutation in Russia. Families from 28 cities came to Moscow, and the LouLou Foundation helped us with tickets and hotels. Many came with other children, with fathers. I really liked that many people have complete families, their fathers do not abandon them, they help and give their wives a rest. And this year, on June 1, we are preparing a second such meeting, 145 people will come to it.
But it was very difficult to find a hotel for our event. The only hotel that agreed to accept our families was the Holiday Inn on Paveletskaya. We sent about 50 letters to different hotels, but they answered us: “We don’t have ramps,” “We are overcrowded.” In fact, this is not the reason, of course. It’s just that society is not yet ready to see children in serious condition, in wheelchairs.