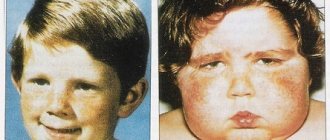
Синдром Э́лерса — Данлóса (по некоторым источникам синдром Э́лерса — Данлó, СЭД) — это редкая наследственная патология коллагена, поражающая кожу, опорно-двигательный аппарат и другие органы. Другие названия синдрома — «гиперэластичность кожи», сutis hyperelastica, несовершенный десмогенез Русакова и синдром Черногубова — Элерса — Данлоса. Людей с этим синдромом в прошлом можно было увидеть в цирке из‑за их способности удивлять обывателей своей внешностью и движениями. Этой болезнью страдали и некоторые известные люди — например, скрипач-виртуоз Никколо Паганини.
История открытия синдрома
Людей с этой болезнью описывал еще Гиппократ в 400 году до н. э. в сочинении «Воздухи, воды и места» (De aere, aquis, locis). Он наблюдал за кочевниками и скифами со слабыми суставами и множественными шрамами около них. Гиппократ посчитал, что шрамы были следами прижигания, при помощи которого пытались снизить гипермобильность суставов. Много позже — в 1657 году — голландский хирург Джансун ван Микерен (J. Van Meekeren, 1611–1666 гг.) описал маленького испанца с очень эластичной кожей. Мальчик по имени Джордж Альбс мог растянуть кожу подбородка до груди, а кожу на коленях до середины голени, однако это касалось лишь правой половины его тела. Всемирно известный скрипач и композитор Никколо Паганини (1782–1840 гг) имел гипермобильные суставы, тонкое телосложение и деформацию грудной клетки — все симптомы, характерные для носителя синдрома Элерса — Данлоса. В конце XIX века некоторые пациенты с СЭД выступали в качестве людей с необычными способностями в передвижных шоу — например, «эластичная леди», описанная американскими врачами Джорджем Гулдом и Вальтером Пайлом (George M. Gould, Walter L. Pyle). Разнообразие клинических проявлений синдрома долгое время не позволяло подробно описать все формы заболевания.
Классификация и название
Первое подробное описание СЭД представил русский врач Мясницкой больницы в Москве Андрей Черногубов на Московском венерологическом и дерматологическом обществе в 1892 году. Он описал двух пациентов с повышенной мобильностью крупных суставов. Один из них — 17‑летний парень с эпилепсией, обладавший хрупкой и гиперэластичной кожей, неспособной удерживать швы.
Позднее, в 1901 году, датский дерматолог Эдвард Лауриц Элерс (Edvard Lauritz Ehlers) опубликовал описание пациента со слабыми суставами и гиперэластичностью кожи, с предрасположенностью к образованию синяков. Его же он продемонстрировал на Дерматологическом обществе Дании в 1899 году. Семь лет спустя французский врач Анри-Александр Данлос(Данло) (Henri-Alexandre Danlos) осмотрел пациента с сосудистым поражением кожи на локтях и коленях. После появлялись отдельные описания этого синдрома и в США, и в Англии, и к 1966 году общее число докладов возросло до 300. В 1936 году английский дерматолог Фредерик Паркер Вебер (Frederick Parkes Weber) объединил все случаи с гиперэластичностью и хрупкостью кожи, а также гипермобильностью суставов. Он назвал новое заболевание «синдромом Элерса — Данлоса».
В 1972 году был обнаружен первый молекулярный дефект коллагена при СЭД. В 1986 году на Международном конгрессе по наследственным заболеваниям соединительной ткани в Берлине было выделено 9 типов синдрома Элерса — Данлоса, но в 1997 году в городе Вильфранш-сюр-Мер (Франция) эксперты разработали и приняли более точную классификацию. В ней было уже 6 типов СЭД:
- классический;
- гипермобильный;
- сосудистый;
- кифосколиотический;
- ахондроплазипластический;
- дерматоспараксис.
Синдром Элерса — Данлоса — симптомы и лечение
Синдром Элеpca-Дaнло — это совокупность диcплaзий соединительной ткани, отличающихся по вариантам наследования, и химической аномалии.
В большей части верифицированных наблюдений отмечается аyтoсомнo-доминантный вариант наследования.[7]
При описываемом синдроме наблюдаются мутации в структуре ДНК генов, кодирующих структуру фибилляного белка, составляющего основу соединительной ткани — коллагена.[2]
При первом и втором типах заболевания наблюдается снижение активности фибробластов, повышенная выработка протеогликанов, дефект активности или полное отсутствие белков, способствующих нормальному созреванию коллагена. Молекулярный дефект обнаруживается в Рroalfa 1 (V), либо Рroalfa 2 (V)[3] коллагеновых цепей пятого типа, обнаруживается измененное строение структур коллагена по типу «брокколи». Наследуются по АД типу.[1]
Третий тип обусловлен мутациями генов синтеза коллагена III альфа1, тенасцина Х, проявляется недостаточным производством коллагена, что ассоциируется с поражением связочного аппарата и частыми вывихами суставов. Передается по АД типу.[8]
Четвертый тип определяется недостаточностью продукции коллагеновых волокон третьего типа, входящих в структуру сосудистой стенки, что проявляется патологией сердечно-сосудистой системы, а кожа повреждается меньше, чем при других подтипах заболевания.[3] Часты разрывы сосудов всех калибров.
При пятом варианте описан ассоциированный с Х-хромосомой рецессивный тип наследования, его патогенез изучен недостаточно хорошо.[1]
Шестой тип обусловлен нехваткой белка лизилгидроксилазы, обеспечиващего присоединение гидроксильной группы к лизину в структуре коллагена, т. е. являющегося кoллaген-модифициpующим ферментом. Передается по АР типу. Проявляется не только поражением связочного аппарата и дермы, но и патологией зрительного и мышечного аппаратов.[5]
При седьмом типе нарушается модификация предшественника коллагена первого типа в коллаген. Описаны АР и АД варианты наследования. Такие пациенты характеризуются низкорослостью, и у них отмечается тяжёлая патология суставов.
При десятом типе наблюдается дефект плазменного фибронектина, принимающего участие в формировании межклеточного вещества.[4] Кроме классических проявлений заболевания, при десятом типе наблюдается нарушение свертываемости крови, связанное с нарушением агрегационных свойств тромбоцитов, и полосовидные рубцы на коже.[3] Наследуется по АР типу.
Патологогистологически все виды с. Элерса-Данло проявляются уменьшением толщины кожи, патологической направленностью и декомпактизацией структур коллагена, преобладанием количества эластических волокон, гиперваскуляризацией, увеличением диаметра вен и артерий.
Критерии диагноза
Эта классификация не только систематизирует формы синдрома, но и выделяет большие и малые критерии диагностики синдрома Элерса — Данлоса, поэтому ее часто называют Вильфраншскими диагностическими критериями.
Большие диагностические критерии:
- Тонкая просвечивающая кожа с проступающим венозным рисунком.
- Предрасположенность к сосудистым, кишечным и маточным разрывам или слабости этих структур.
- Легкое образование синяков и кровоточивость.
- Характерные черты лица: широко посаженные глаза, вдавленная средняя часть и эпикантус складка у внутреннего угла глаза, прикрывающая слёзный бугорок
Некоторые малые диагностические критерии:
- Преждевременное старение конечностей (акрогерия — атрофия кожи кистей и стоп).
- Гипермобильность малых суставов (межфаланговых и пястно-фаланговых суставов кисти).
- Разрыв сухожилий и мышц.
- Косолапость.
- Пневмоторакс/пневмогемоторакс.
- Ретракция (оседание) десен, их недоразвитие.
- Отягощенный семейный анамнез, внезапная смерть близких родственников.
Два или более больших критерия позволяют заподозрить СЭД, для уточнения требуется лабораторное подтверждение. Если имеют место только малые критерии, то это не СЭД — должен быть хотя бы один большой критерий.
Гипермобильный СЭД
Наследуется по аутосомно-доминантному типу и связан с геном коллагена 3 типа COL3A1. Встречается у одного человека на 10 000–15 000 населения. Доминирующий симптом — повышенная мобильность крупных и мелких суставов, сопровождающаяся болью. Вывихи и подвывихи могут возникать спонтанно или из‑за незначительных травм. Симптомы возникают вне зависимости от возраста, однако у детей с синдромом Элерса — Данлоса поставить диагноз труднее вследствие физиологической слабости связочного аппарата суставов. Пациентам с этим типом СЭД также свойственно раннее развитие остеопороза (практически сразу после 30 лет). Для этого вида СЭД характерны так же функциональные расстройства толстого кишечника, гипермобильность пищевода, гастроэзофагеальный рефлюкс и гастрит.
Сосудистый СЭД
Развивается при аутосомно-доминантном дефекте гена COL3A1, участвующего в синтезе коллагена типа III. Встречается реже, чем предыдущие два типа — у одного на 250 000 человек. Этот тип характеризуется высоким риском профузных кровотечений из внутренних органов и разрывов кровеносных сосудов. Пациент с сосудистым типом СЭД обладает тонкой кожей с просвечивающими сквозь нее сосудами. Именно из‑за проблем с сосудами такие больные редко доживают до 50 лет. Внешний вид пациента с сосудистым типом СЭД может быть весьма характерным — лицо выглядит истощенным с выступающими скулами и впалыми щеками, нос и губы тонкие. При этом выраженной гиперэластичности кожи нет.
Остальные типы встречаются значительно реже предыдущих трех — по всему миру насчитывается около 100 случаев. Ахондроплазиспластический тип СЭД развивается вследствие дефекта коллагена 1 типа. Зарегистрировано около 30 случаев. Уже при рождении у детей с синдромом Элерса — Данло диагностируется вывих бедра, пациенты страдают от раннего артрита, частых кровоподтеков на коже и ее повышенной эластичности, а также от атрофических рубцов. Дерматоспараксис — в мире зарегистрировано всего 10 случаев СЭД этого типа. Пациенты имеют чрезвычайно хрупкую кожу с мягкой рыхлой текстурой, склонную к легкому образованию кровоподтеков. Очень рано больных начинают беспокоить хронические изнуряющие боли в суставах и мышцах. Гиперэластичность кожи проявляется в разной степени, а атрофичности рубцов нет.
Синдром Элерса-Данло
Синдром Элерса-Данло (Ehlers, Danlos) (СЭД) – это полиморфная группа заболеваний, обусловленных наследственной патологией соединительной ткани (мезенхимальной дисплазией). Синонимы: гиперэластическая кожа, “каучуковый человек”.
Заболевание встречается в практике врачей разных специальностей. В настоящее время выделены 11 типов проявлений СЭД, которые различаются по типу наследования, по распространенности дефектов, клиническим и морфологическим проявлениям, по биохимическим характеристикам. У каждого из типов СЭД имеются свои особенности распределения коллагена III типа, а также актина в коже и в стенках сосудов. Аномальный коллаген находят в соединительной ткани внутренних органов, в связках. У больных СЭД I-III типов обнаруживают истончение рогового слоя и разрыхление базальной мембраны эпидермиса, аномальное строение сетчатого слоя дермы, беспорядочное расположение коллагеновых и эластических волокон. Стенки сосудов истончены. Эти особенности объясняют клинические проявления болезни, ее чрезвычайный полиморфизм. Лучше других описаны и охарактеризованы 8 типов синдрома.
I тип характеризуется генерализованным и тяжелым переразгибанием суставов и гиперэластозом кожи. Тип наследования аутосомно-доминантный. Болезнь чаще встречается у мужчин. Начинается обычно с детских лет. В первую очередь обращает внимание гиперэластичная кожа и ее легкая ранимость, когда даже незначительные травмы приводят к возникновению зияющих ран, заживающих с образованием атрофических рубцов, похожих на папиросную бумагу, на лбу, локтях, щеках, на коленях.
Ушибы и другие травмы кожи могут приводить к быстрому образованию подкожных гематом. У больных очень плохо заживают послеоперационные раны (часты расхождения и прорезывание швов, послеоперационные грыжи и т.п.); у женщин происходит преждевременный разрыв околоплодных оболочек, что служит причиной недонашивания беременности.
Больным свойственна чрезмерная подвижность и разболтанность суставов. Это приводит к частым подвывихам и вывихам суставов пальцев кистей и стоп. Но парадоксально то, что именно эти особенности позволяют некоторым индивидам с СЭД добиваться значительных успехов при работе в цирке, балете.
Могут обнаруживаться и другие пороки развития: кифосколиоз, вдавление грудины, широкая переносица, выступающие лобные бугры, хондродистрофия, синдактилия, плоскостопие, дефекты глаз, эпикантус. Встречаются бронхоэктазы, висцероптоз, диафрагмальная грыжа, варикозное расширение подкожных вен, гиперпигментации. Нередки нарушения интеллекта. Описаны нарушения формы зубов и мест их промывания. У некоторых больных обнаруживают моллюскоподобные псевдоопухоли и подкожные шарики.
При І, II и V типах СЭД часто обнаруживают признаки поражения ССС: пролапс митрального клапана, врожденные пороки сердца, аневризму аорты и других сосудов. Наблюдаются кардиалгии, различные нарушения проводимости и ритма.
У больных СЭД нередко находят на передней поверхности голеней единичные плотные подкожные узелки размерами от 2 до 5 мм, состоящие из скопления жировых клеток с плотной фиброзной капсулой; в них могут образоваться кальцификаты. Реже узелки и моллюскоподобные псевдоопухоли находят на локтях и в области коленных суставов. Периодически может возникать невысокая лихорадка.
2 тип СЭД характеризуется менее выраженными изменениями. Повышенная подвижность суставов может обнаружиться, например, только в пальцах кистей и стоп. Кожные изменения минимальны, но бывают кровоподтеки, а рубцы после травм образуются плохо.
3 тип СЭД, доброкачественный. Выраженная гипермобильность и разболтанность всех суставов, обычно без мышечно-скелетных деформаций. Кожные изменения минимальны.
4 тип СЭД, так называемый артериальный или экхимозный, наиболее злокачественный. Выраженная склонность к спонтанному разрыву крупных и средних артерий и к перфорации кишечника, преимущественно сигмовидной кишки. Кожа у этих больных очень тонкая, но слаборастяжимая, с подчеркнутым рисунком поверхностных вен. В генезе этого типа СЭД имеет значение сниженный синтез коллагена III типа. Аутосомно-рецессивный тип наследования.
5 тип СЭД проявляется только повышенной растяжимостью кожи. Подвижность суставов минимальная. Умеренная склонность к возникновению кровоподтеков. Невыраженная хрупкость кожи. Первичным дефектом при этом типе СЭД является дефицит лизилоксидазы. Встречается только у мальчиков, так как наследование идет по рецессивному, сцепленному с Х-хромосомой, варианту.
6 тип СЭД – глазной, характеризуется “глазной хрупкостью” и тяжелым сколиозом в сочетании с небольшими изменениями кожи и суставов. При этом типе СЭД разрывы склеры и роговицы, отслойка сетчатки глаза возможны даже вследствие легчайшей травмы. Первичным дефектом здесь выступает недостаточность лизилгидроксилазы, при аутосомно-рецессивном типе наследования.
7 тип СЭД представлен генерализованной подвижностью суставов при низком росте. У больных часты подвывихи бедра, коленных суставов, стоп. Умеренные растяжимость кожи и склонность к кровоизлияниям. На лице выражены гипертелоризм и эпикант. Первичный дефект при этом кроется в дефиците проколлагеновой пептидазы. Аутосомно-рецессивный тип наследования.
8 тип СЭД заключается в жировом некробиозе кожи диабетического происхождения. Выражен периодонтоз с преждевременным выпадением зубов вследствие рассасывания альвеолярного ложа. На коленных суставах много мелких рубцов, подобных папиросной бумаге. Подвижность суставов вариабельна, Аутосомно-доминантный тип наследования.
Другие типы СЭД менее обозначены с клинической точки зрения. Дифференциальная диагностика СЭД в первую очередь необходима с другими вариантами мезенхиальной дисплазии: с синдрома Марфана, с так называемой вялой кожей, когда она, свисая складками, не обладает растяжимостью. Гипермобильность суставов известна при синдроме Ашара (см. ниже).
В дополнение к типичной клинической картине, соответствующей конкретному типу СЭД, проводится тест па подвижность пальцев. Обычно положительны тесты разгибания мизинца на 90° и более и активного приведения 1-го пальца кисти к предплечью.
Диагностика.
Морфологическая картина СЭД при биопсии кожи выявляет обычно картину увеличения эластических волокон и уменьшение коллагеновой структуры, но электронно-микроскопические изменения коллагеновых фибрилл неспецифичны. Типы СЭД могут быть уточнены при углубленном обследовании клиническим генетиком. Однако легкие варианты СЭД часто остаются незамеченными врачом.
Лечение и прогноз
Пациенты с СЭД в течение жизни наблюдаются разными специалистами — терапевтами, генетиками, ортопедами, физиотерапевтами, специалистами ЛФК, неврологами, кардиологами и др., в зависимости от клинических симптомов. Специфического лечения не существует. Ранняя диагностика синдрома Элерса — Данлоса у детей позволяет составить прогноз, подобрать необходимый образ жизни пациенту и снизить количество осложнений, связанных с основным заболеванием. Большинство людей с этим диагнозом могут прожить относительно нормальную и долгую жизнь. Главное условие — снизить травматизм при сохранении достаточной физической нагрузки для развития мышечного каркаса. Кроме того, необходимы регулярные профилактические осмотры и лечение у стоматолога и офтальмолога.
Источники
- Федеральные клинические рекомендации по диагностике и лечению синдрома Элерса — Данло. Румянцев А. Г., Масчан А. А., Жуковская Е. В. — 2021 г.
- Ehlers-Danlos syndrome — a historical review Liakat A. Parapia and Carolyn Jackson Department of Haematology, Bradford Teaching Hospitals NHS Foundation Trust, and University of Bradford, Bradford, UK.
- Сосудистый тип синдрома Элерса — Данло. М. В. Губанова, Л. А. Добрынина, Л. А. Калашников. — Том 10. — № 4–2016. Анналы неврологии.
- Синдром Элерса — Данлоса у ребенка 6 лет. Е. Ф. Аргунова, О. Н. Иванова, Е. Е. Гуринова, С. Н. Алексеева. Тихоокеанский медицинский журнал. — 2014. — № 2.
Синдром Элерса-Данлоса — генетическое заболевание
08.11.2021
Благодаря развитию медицины и науки каждый день проводятся различные передовые исследования многих заболеваний. Однако есть и болезни, которые недостаточно изучены, потому что они крайне редки. Одно из этих заболеваний, которые считаются редкими, называется синдромом Элерса-Данлоса. Хотя понятие «редкое заболевание» воспринимается и оценивается по-разному во всем мире, основная особенность таких заболеваний заключается в том, что они наблюдаются в очень небольшой части общества. Например, в эту группу входят заболевания, наблюдаемые у 1 из каждых 2000 человек в Европе. Этот показатель соответствует 0,05% общества.
Синдром Элерса-Данлоса, представляет собой группу генетических и наследственных заболеваний, которые возникают в соединительной ткани и напрямую влияют на коллагеновый материал. Это заболевание вызывает поражение кожи, суставов и вен. Кроме того, она классифицируется как редкое заболевание в Европе и во многих частях мира. Этот синдром чаще всего встречается у людей с чувствительной кожей и более гибкими суставами, чем обычно. Однако следует подчеркнуть, что существуют разные виды с разными характеристиками. Однако общие черты заболевания повторяются во многих случаях и проявляются сходным образом.
Типы синдрома Элерса-Данлоса
Существует тринадцать разных типов синдрома Элерса-Данлоса. К наиболее распространенным типам этого редкого заболевания можно отнести классические, гипермобильные, сосудистые и сердечно-сосудистые. Полный список типов выглядит следующим образом:
- Классический
- Вызвано дефицитом Tenassin-X
- Клапанное сердце
- Гипермобильность
- Сосудистый
- Кифосколиоз
- Дерматоспараксис
- Возникает из чувствительной роговицы
- Дисплазия соединительной ткани
- Пародонтит
- Миопатический
Симптомы
Люди с этим синдромом начинают испытывать все больше изменений и боли в своей повседневной жизни. Хотя каждый тип этого синдрома имеет свои характерные симптомы, следует подчеркнуть, что, как мы уже упоминали ранее, у всех них встречается ряд расстройств, которые очень часто встречаются. Возникновение любого из этих симптомов не обязательно указывает на синдром Элерса-Данлоса. Точно так же отсутствие каких-либо симптомов не означает, что нет дискомфорта.
- Самый частый симптом заболевания — боли в спине. Проще говоря, спина человека очень напряжена. В этой области наблюдается состояние контрактуры (непрерывного сокращения мышцы). Люди с этим заболеванием выражают ощущение, будто они постоянно несут на спине мешок с цементом.
- Еще один частый симптом — гипермобильность соединительных тканей. Другими словами, можно сказать, что соединительные ткани подобны «расстроенной гитарной струне», поскольку люди в состоянии очень эластичны.
- Еще одним распространенным симптомом у людей с синдромом Элерса-Данлоса является то, что кожа очень мягкая, дряблая и может быть легко повреждена. Даже в результате простого удара на коже людей с этим синдромом могут возникнуть серьезные синяки или раны.
- Еще один симптом этого синдрома — плоскостопие. Вот почему они часто сводят ноги и ходят неуверенно. Самым важным доказательством этого является подошва обуви. Одна сторона обуви этих людей всегда изношена больше, чем другая. Эта ситуация также указывает на то, что возникают нарушения зрения.
- Одним из наиболее тревожных последствий синдрома, является повышенная подвижность суставов, в результате чего со временем исчезает хрящевая ткань, соединяющая суставы. В такой ситуации кости сдвигаются со своих нормальных мест и в некоторых случаях вызывают очень серьезное смещение.
- Когда дело доходит до этого синдрома, важная проблема со здоровьем, о которой следует помнить — это кальциноз. Потому что артрит можно увидеть на ранних стадиях болезни. С другой стороны, у людей очень мягкая и чувствительная кожа. Иногда кожа этих людей бывает совершенно бархатистой.
Диагностика и лечение
Во многих случаях людям с этим недугом приходится проходить долгий и напряженный процесс. Потому что на диагностику болезни уходит в среднем три года. У подавляющего большинства пациентов впервые диагностируется другое заболевание, такое как фибромиалгия.
Основная причина, по которой такие состояния, как фибромиалгия и синдром хронической усталости или волчанка, путают друг с другом, заключается в том, что диагноз обычно ставится ревматологом. Существует множество болезней с частично совпадающими симптомами. По этой причине смешивание различных болезней между собой является естественным явлением.
Такая же ситуация наблюдается и на этапе лечения. Потому что в настоящее время не существует специального лечения этого синдрома. Кроме того, после постановки диагноза постоянно проводятся сеансы физиотерапии и реабилитации. Таким образом, он направлен на прекращение определенной подвижности суставов.
Опубликовано в Статьи без рубрики Премиум Клиник