Systemic scleroderma (according to ICD 10 - progressive systemic sclerosis, code M34.0) is an autoimmune disease of connective tissue. Its main clinical manifestations are associated with widespread ischemic disorders due to obliterating microangiopathy, fibrosis of the skin and internal organs (heart, lungs, kidneys, digestive and peripheral nervous systems), and damage to the musculoskeletal system. The Yusupov Hospital has created all the conditions for the treatment of patients suffering from systemic scleroderma:
- Comfortable living conditions in the wards;
- Diagnosis of the disease using modern equipment and the latest laboratory research methods;
- Treatment with the most effective drugs that have a minimal range of side effects;
- Attentive attitude of medical staff.
Women suffer from systemic scleroderma 3-7 times more often than men. The onset of the disease is possible at any age, but people aged 30 to 60 years are more likely to get sick.
Causes and mechanisms of development of systemic scleroderma
The exact causes of systemic scleroderma have not been established today. Scientists suggest that the disease develops under the influence of many external and internal factors combined with a genetic predisposition to the disease. Along with the role of infection, vibration, cooling, stress, injury and endocrine disorders, special attention is paid to the effects of chemical agents (household, nutritional, industrial) and certain medications.
The central links in the mechanism of development of systemic scleroderma are excessive fibrosis, disorders of the immune system and microcirculation. An imbalance of cellular and humoral immunity leads to activation of the production of specific antinuclear antibodies, interleukins (mediators of inflammation and immunity), antibodies to the endothelium and connective tissue.
In the inner layer of the vascular wall, the number of smooth muscle cells increases and mucoid swelling occurs. The lumen of the vessels narrows, microthrombosis develops. Over time, blood supply to tissues decreases. The degree of endothelial damage is indicated by an increase in the concentration of von Willebrand factor and cellular soluble adhesion molecules.
T-lymphocytes and fibroblasts accumulate in the dermis (the skin itself). They overproduce collagen types I and III. Activated mast cells produce transforming growth factor B and histamine. This stimulates the proliferation of fibroblasts and the formation of components of the intercellular matrix.
Types of systemic scleroderma
There are diffuse and limited clinical forms of systemic scleroderma. In 2% of cases, scleroderma without scleroderma is registered (damage to internal organs only). There are cross-forms of the disease in which systemic scleroderma is combined with rheumatoid arthritis, inflammatory myopathies, and systemic lupus erythematosus).
The following variants of the clinical course of scleroderma are distinguished:
- Acute, rapidly progressive course - generalized fibrosis of the skin (diffuse form) and internal organs develops during the first two years from the onset of the disease;
- Subacute, moderately progressive course - there are clinical and laboratory signs of immune inflammation (dense swelling of the skin, myositis, arthritis), there may be cross-syndromes;
- Chronic, slowly progressive course - vascular pathology predominates; at the beginning of the disease, Raynaud's syndrome is observed for a long time, then the skin and internal organs are gradually affected.
Causes
The exact causes of scleroderma have not yet been determined. Experts identify predisposing factors, the presence of which increases the risk of autoimmune disease. These include:
- Genetic predisposition. The presence of scleroderma in relatives significantly increases the likelihood of developing the disease in future generations. There is a theory according to which a defect in the HLA-9QA1 gene is responsible for the formation of an autoimmune disease. Genome mutations can be congenital or acquired. The following factors contribute to the development of a genetic defect:
- Radiation exposure.
- Prolonged exposure to ultraviolet rays.
- Taking certain medications. For example, cytostatics.
- Viral or bacterial infection.
- Exposure to high and low temperatures.
- Inflammatory process. Biologically active substances produced during the inflammatory reaction can provoke tissue and vascular changes. Vascular permeability increases, and the inflammatory process intensifies. As a result, the prerequisites are created for the formation of chronic inflammation. The risk of scleroderma increases with the addition of other provoking factors.
- Immune disorders. Scleroderma is an autoimmune disease in which the body's own cells are perceived as foreign. Immune reactions are triggered by connective tissue growth factor.
- Infectious diseases. There is no consensus regarding the involvement of infectious agents in the development of scleroderma. Experts do not identify a specific pathogen that provokes autoimmune processes. It was noticed that in some patients with diagnosed scleroderma, cytomegalovirus fragments were detected.
- Environmental factors. Experts highlight a theory according to which there is a relationship between exposure to environmental factors and the occurrence of scleroderma. If there is a genetic predisposition, exposure to environmental factors increases the risk of developing an autoimmune disease. The main provoking factors include:
- Chemical compounds. For example, silicon, mercury, benzene, toluene, epoxy resin.
- Nutritional supplements. Among them are L-tryptophan, mazindol, fenfluramine.
The mechanism of scleroderma occurrence is associated with the development of an immune response against the body's own cells. As a result, an inflammatory process is formed, thinning the skin and blood vessels. Circulating complexes settle on internal organs, causing their damage. Scleroderma is accompanied by the formation of sclerotic changes with the formation of scar tissue.
Symptoms of systemic scleroderma
The clinical picture of systemic scleroderma includes a wide range of manifestations. It is characterized by polymorphism in terms of symptoms, severity and prognosis of the disease. One of the first symptoms of systemic scleroderma is a syndrome of symmetrical paroxysmal spasm of arteries, precapillary arterioles, cutaneous arteriovenous anastomoses under the influence of cold and emotional stress.
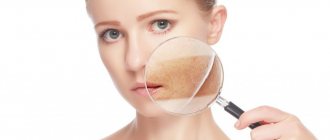
Skin lesions in systemic scleroderma are characterized by stages. In the initial stages of the disease, dense swelling of the skin and underlying tissues occurs. Due to swelling of the fingers, the hand is difficult to clench into a fist in the morning. Sometimes diffuse hyperpigmentation develops at the onset of the disease.
As the disease progresses, skin induration occurs - an increase in density and a decrease in elasticity. The skin can hardly be gathered into a fold. It is much thicker than normal skin. At the atrophy stage, the skin acquires a bluish-brown color due to hyperpigmentation and dyspigmentation, and becomes thinner. It has a characteristic shine. The skin becomes rough, dry, hair disappears. Small subcutaneous and intradermal deposits of calcium salts may appear in soft tissues. If they open, a cheesy mass is released and long-term non-healing ulcers are formed.
In the advanced stage of the disease, the “purse string” symptom appears - when radial folds form around the mouth. Late signs of systemic scleroderma are telangiectasia - dilated venules and capillaries grouped in a bundle. The number of telangiectasias increases over time. They are located on the hands, face, and décolleté. These symptoms of systemic scleroderma are visible in the photo.
Damage to the musculoskeletal system is manifested by polyarthralgia; polyarthritis with severe synovitis and tenosynovitis may develop. In the chronic course of systemic scleroderma, sclerosing tenosynovitis and damage to other tissues located around the joints predominate. These processes, together with skin thickening, lead to the formation of sclerodactyly (local thickening and hardening of the skin of the fingers), flexion contractures of small and large joints. A typical manifestation of systemic scleroderma is acroosteolysis - resorption of the terminal sections of the terminal phalanges of the hands due to prolonged ischemia.
The manifestations of scleroderma are the following lesions of internal organs:
- Hypotension of the esophagus;
- Erosion and ulcers of the esophagus;
- Damage to the small and large intestines;
- Malabsorption syndrome;
- Interstitial lung disease;
- Pulmonary hypertension;
- Acute or chronic nephropathy.
With scleroderma damage to the heart, fibrosis develops involving both ventricles. In patients, the balance of the immune system is disturbed, autoimmune disorders occur with the formation of antinuclear antibodies. Almost all patients have antinuclear factor.
The most important blood test for systemic scleroderma is the detection of anticentromere antibodies (ACAs), antibodies to topoisomerase 1 (Scl-70) and to RNA polymerase III. All of these autoantibodies are directed to soluble nuclear proteins. Autoantibodies in systemic scleroderma appear already at the preclinical stage. Each person with systemic scleroderma usually has only one type of autoantibody, which does not change as the disease progresses.
Scleroderma signs, photos
There are two main forms of focal scleroderma: plaque and linear. Sometimes a generalized form is noted.
The plaque form of focal scleroderma manifests itself in the form of one or more limited, compacted spots or plaques, usually with hypo- or hyperpigmentation.
The generalized form is a more severe version of the disease, which is characterized by many foci, often merging and occupying a large surface area of the body.
A combination of several forms of the disease is possible, but from the point of view of treatment approaches, these varieties are not very different from each other.
Patients may complain of itching, tingling, pain, a feeling of “tightness” of the skin in the affected areas, and limited movement in the joints.
Diagnosis of systemic scleroderma
Rheumatologists at the Yusupov Hospital make a diagnosis of systemic scleroderma based on the clinical picture of the disease, data from instrumental and laboratory studies, which allow one to assess the degree of involvement of internal organs in the pathological process and the severity of pulmonary hypertension. For this purpose, I use the following research methods:
- Electrocardiography;
- Echocardiography;
- Six-minute walking test;
- X-ray and computed tomography of the chest;
- Spirometry;
- Body plethysmography;
- Ventilation-perfusion lung scintigraphy;
- Angiopulmonography.
If indicated, catheterization of the right heart is performed. Patients are prescribed a clinical, biochemical, immunological blood test, and a coagulogram. In a general blood test for systemic scleroderma, an increase in the erythrocyte sedimentation rate, a change in the amount of hemoglobin and red blood cells can be detected. A general urine test reveals hyposthenuria (decreased urine density), proteinuria (protein in the urine), decreased specific gravity of urine, and casts. A biochemical blood test includes determination of the level of AlT, AST, total protein and fractions, glucose, creatinine, urea, and cholesterol. In the blood of a patient suffering from systemic scleroderma, an increase in the titer of the following antibodies is detected:
- Antinuclear;
- Anticentromeric;
- To topoisomerase-1 (Scl-70).
Rheumatologists use clinical guidelines from the American Rheumatological Association to make a diagnosis of systemic scleroderma. The “major” criterion is proximal scleroderma. It is characterized by symmetrical thickening, thickening, and induration of the skin proximal to the metacarpophalangeal and metatarsophalangeal joints. Changes characteristic of scleroderma can be located on the neck, face, chest, and abdomen. “Minor” criteria include sclerodactyly (the skin changes listed above are limited to the fingers), small scars on the fingertips or loss of fingertip tissue, and bilateral basal pneumofibrosis on radiographic examination. The diagnosis of systemic scleroderma is established in the presence of a “major” criterion or two “minor” criteria. Rheumatologists use these criteria to identify severe systemic scleroderma. They do not cover early limited, crossed and visceral systemic scleroderma.
Treatment of scleroderma in Moscow
Autoimmune diseases require timely diagnosis and correct treatment. You can get examined in Moscow at the Yusupov Hospital. The clinic has been identifying and treating any rheumatological diseases for many years. The hospital has the latest equipment that allows us to carry out the diagnostic measures necessary to clarify the presence of scleroderma. Based on the data obtained, highly qualified personnel conduct a course of individual treatment, selected in accordance with the stage of the disease. You can make an appointment with a specialist, as well as ask any question you have by phone or on the official website of the Yusupov Hospital.