One of the causes of female infertility may be Shereshevsky-Turner syndrome. This is a genetic disease caused by the absence of the second X chromosome. We can say that it is she who finally makes a woman out of a woman.
Natural pregnancy with Shereshevsky-Turner syndrome is practically impossible, since the sick woman does not produce eggs of the required quality. Previously, such a diagnosis became a death sentence, and the patient lost all hope of having a child. Today, science has stepped forward, and after undergoing certain treatment, such women can experience the happiness of motherhood. The IVF Center clinic offers you an almost win-win method on how to get pregnant with a diagnosis of Shereshevsky-Turner syndrome - IVF. It is preferable because it reduces the risk of transmitting the disease to offspring.
Shereshevsky-Turner syndrome: characteristics of the disease
This disease was discovered by N.A. Shereshevsky almost 90 years ago. The Soviet endocrinologist gave a detailed description of the pathology, suggesting that the pituitary gland and gonads, which do not fully perform their functions, are responsible for its manifestation. The scientist also drew attention to congenital malformations of the internal development of patients. More than ten years later, Turner described the syndrome somewhat differently and outlined its main external symptoms.
The chromosomal nature of the disease was discovered by Charles Ford in the late fifties, but his name is not reflected in the name of the disease. Subsequently, it was proven that monosomy plays a decisive role in the development of a number of anomalies of the body described by Ford’s predecessors. There has been a long debate in science about who is the discoverer of the disease.
So, Shereshevsky-Turner syndrome, the causes of which are the absence of the X chromosome or its replacement with an isochromosome, occurs in one newborn girl out of four thousand. Often pregnancies with fetuses with this pathology end in miscarriages.
If a child is diagnosed with Shereshevsky-Turner syndrome during intrauterine development, the baby's karyotype will be 45X0. It is noteworthy that initially the embryo develops normally, and only at the time of birth the germ cells undergo atresia. In a newborn, the ovaries will be rudimentary, or there will be no follicles in them.
Lymphedema
In the photo, lymphedema - due to a violation of the outflow of lymph and its accumulation in the intercellular space, the volume of the limbs increases.
Lymphedema is a disease in which swelling of a part of the body appears, which occurs due to improper functioning of the lymphatic system (see photo of lymphedema).
Lymphedema is a chronic pathology of the lymphatic system, leading to other diseases such as ulcers, increased pigmentation, eczema, thickening, etc.
Shereshevsky-Turner syndrome: mosaic form
In the mosaic form of this disease, two types of cells are combined in the patient’s body. Some have a normal karyotype, while others show pathology. In general, the karyotype will look like 46XX/45X. The woman’s health status will depend on the proportion of cells with a normal karyotype and cells without one X chromosome.
Many patients develop sexual characteristics, including genitalia. The chances of pregnancy with the mosaic type of syndrome are much higher, and conception can even occur naturally. However, a pregnant woman with this diagnosis will need prenatal karyotyping, since the fetus will be at risk.
If the expectant mother has Shereshevsky-Turner syndrome, prevention of it in the child is mandatory. It consists of identifying pathologies of intrauterine development and consulting with a geneticist.
Diagnostics
Shereshevsky-Turner syndrome in newborn girls allows one to suspect the presence of lymphedema or a pterygoid fold in the neck. If such signs are absent, then suspicion of the presence of this pathology may appear later due to short stature, amenorrhea, and also in the absence of puberty. A karyotype study allows you to confirm or refute such a diagnosis. In general, the diagnosis can be established by the presence of a clinical picture characteristic of this disease.
MRI or echocardiography is performed to identify congenital heart defects. Some specific examinations are also carried out, in particular, cytogenetic research . A blood test during the development of this disease demonstrates a decrease in the amount of estrogens and at the same time a high content of pituitary . X-ray examination often reveals osteoporosis , as well as pathologies of the development of the bone skeleton. In addition, during the diagnostic process, an examination is carried out aimed at identifying concomitant diseases.
Also in modern medicine, screening tests , which make it possible to find out about the risk of a fetus having Shereshevsky-Turner syndrome.
Shereshevsky-Turner syndrome: symptoms of pathology
The time of onset of the disease is not the same in all patients. For some, it is diagnosed in the prenatal period. Such children are born with a body length of no more than 48 cm and a weight of 2500-2800 kg. In others, the pathology manifests itself after a few years: the girl is noticeably stunted in growth, parents are struck by a valgus deviation in the elbow joints, a low hairline on the back of the neck, and drooping eyelids. In addition, children with Shereshevsky-Turner syndrome may suffer from mental development delays.
Although this disease is associated with sexual underdevelopment, many of its symptoms are external:
- small height (135-145 cm for an adult woman);
- short neck;
- incorrect physique;
- unusual shape of the chest (from shield-shaped to barrel-shaped);
- shortened wrist bones;
- excess skin on the neck (the so-called “wings”)
- low-set ears, deformation of the ears;
- drooping eyelids, presence of epicanthus;
- an abundance of pigment spots.
This is what people with Shereshevsky-Turner syndrome look like. They usually suffer from cardiac abnormalities, circulatory system defects, and kidney problems. However, it cannot be said that when this diagnosis is made, the child will have a complete set of all of the above. Manifestations for each patient are individual. Turner-Shereshevsky syndrome, the symptoms of which cannot be the same in two women, is remarkable for this. Although statistics provide an approximate picture of the disease, it is almost impossible to find several identical cases.
Clinical manifestations of Shereshevsky-Turner syndrome were first described by N.A. Shereshevsky in 1925 and, independently, by N. Turner in 1928, and chromosomal mutations were identified in 1959. Traditionally, patients are consulted by an endocrinologist in connection with short stature, developmental disorders due to deficiency of sex hormones to resolve clinical issues associated with these phenomena. The study of comorbidity is one of the priority areas of medical research in recent years. In connection with the progression of complex pathology of the cardiovascular system, a prognosis for the life of patients in this group is being formed. Observations of them revealed the advisability of cardiac examination and treatment. Let's consider the principles of consultation by a cardiologist for patients with Shereshevsky-Turner syndrome, a genetic disease associated with comorbid pathology.
The prevalence of this syndrome is 25-55 cases per 100,000 women. It is diagnosed during the first 6 weeks in a third of patients, another third in childhood due to pathology identified by a pediatrician, and the remaining cases in puberty.
When examining patients, a complex pathology is revealed: short stature (100%); cranial deformities; micrognathia (60%), high “arched” palate (38%); short neck (40%); low-set, turned ears; webbed neck (25%), low hair growth at the back (42%); multiple pigmented nevi (26%); hypoplastic and/or underdeveloped nipples; shortening of the fourth metacarpal bone (37%); curvature in the elbow joint (47%); lymphatic obstruction, swelling of the arms and legs (22%); hyper-convex, thin and brittle nails (13%); renal anatomical deformities, horseshoe kidneys (40%); primary hypogonadism (96%); Hashimoto's thyroiditis, hypothyroidism (44%); curvature in the knee joints (35%) [1].
Diagnosis of karyotype abnormalities (45,X (45,X/ 46,XX, structural pathology of the X chromosome) is carried out using genetic testing and PCR analysis. Shereshevsky-Turner syndrome is characterized by mosaicism. In this clinical condition, not all human cells contain identical chromosomes: two or more genetically distinct cell populations may be present. The 45x/46xx variant is most often detected, and 45,X/46,Xr(X) is one of the rarest. The search for an abnormal Y chromosome determines the prognosis of for tumor development: gonadoblastoma or dysgerminoma.The latter is associated with a poor prognosis due to metastatic growth [2].
Risk factors for cardiac mortality in patients with Shereshevsky-Turner syndrome are congenital cardiovascular diseases, aortic dissection, and coronary heart disease [3]. The estimated reduction in life expectancy at the age of 1 year is 12.5 years, and for groups aged 20 and 40 years, respectively, 11 and 10 years [4]. At the same time, the highest mortality rate was found among those with karyotypes 45,X and isoXq [5]. When analyzing mortality in different age groups, it turned out that with increasing age, the contribution of cardiovascular and respiratory pathology increased, and the contribution of congenital heart anomalies decreased [6].
Screening for cardiovascular pathology is carried out as follows. When diagnosing the syndrome, all patients undergo an examination by a cardiologist with an examination of congenital heart defects, a comprehensive examination with measurement of blood pressure in all extremities, as well as high-quality visualization of the heart, aortic valve, aortic arch, and pulmonary veins [7]. The study of complex vascular pathology in Shereshevsky-Turner syndrome contributed to the formulation of imaging recommendations: due to the fact that echocardiography is recommended for diagnosis at an early age, MRI should be used in older girls and adults who are able to tolerate the procedure without sedation. Imaging is recommended every 5–10 years, and the modality of repeat testing is nonspecific [8].
It is recommended to conduct annual screening for cardiovascular diseases in adults with appropriate measurement of body weight, height, blood pressure, blood biochemical parameters (glycemia and HbA1c, creatinine, urea, electrolytes, liver transaminases, lipid profile - if it is not changed, then re-monitor at more frequent intervals), general urine analysis and thyroid hormone levels [9].
Along with traditional risk factors for coronary heart disease (arterial hypertension, insulin resistance, diabetes mellitus, dyslipidemia, obesity), there are also specific ones (estrogens deficiency, growth hormone deficiency, hypercoagulability status and thrombosis) [6]. Factors that determine mortality from coronary artery disease are: cardiac anomalies, endothelial dysfunction, obesity, arterial hypertension, hyperlipidemia, insulin resistance, diabetes mellitus and estrogen deficiency [3]. The clinical course of the syndrome is associated with the development of secondary arterial hypertension due to coarctation of the aorta, hypoplasia of the aortic arch, malformation of the renal vessels and secondary renal dysfunction [6]. Associated pathology increases the risk of progression of cardiovascular pathology compared to population groups without genetic pathology. This should be taken into account when planning dispensary activities.
Screening criteria for identifying aortic coarctation based on history are headache, nosebleeds, and weakness in the legs. An objective examination reveals a murmur over the aorta spreading through the bloodstream. During a comprehensive examination of the defect, signs of blood redistribution among large vessels are established in the patient: a difference in blood pressure (>20/10 mm Hg) between the upper and lower extremities or right and left arms, as well as changes in the shape of the aortic shadow during an X-ray examination of the organs chest with verification based on echocardiography and other imaging tests [10].
To diagnose arterial hypertension, existing clinical guidelines should be used, and some features of this clinical group should be taken into account. Within the framework of the concept of vascular aging, one should consider the results of population-based studies in Shereshevsky-Turner syndrome, which demonstrated an increase in systolic and diastolic blood pressure with the age of patients. A correlation has been established between an increase in body mass index and an increase in systolic and diastolic blood pressure [11]. The target body mass index is considered to be no more than 25 kg/m2 [9]. Based on these facts, non-drug methods for correcting blood pressure are as relevant as for other groups of patients with arterial hypertension. If office blood pressure is detected more than 130/80 mm Hg. Art. 24-hour blood pressure monitoring should be performed. The target average daily blood pressure is 130/80 mm Hg. Art. During treatment, the patient is given only advice on lifestyle changes with blood pressure values from 130/80 to 140/90 mmHg. Art. in the absence of signs of progression of cardiac pathology. In the presence of coarctation of the aorta, bicuspid aortic valve and aortic dilatation, drug antihypertensive therapy should be started when the readings are at least 140/90 mmHg. Art. drug therapy should be carried out in the absence of structural changes in the aorta [9]. These recommendations are based on primary data identifying a linear relationship between systolic blood pressure and aortic root diameter (r=0.5; p=0.003) [12]. Arterial hypertension is a risk factor for the development of aortic dissection [3], and the structural background for the development of this emergency situation is dilatation of the ascending aorta, elongation of the aortic arch, coarctation of the aorta, and bicuspid aortic valve [8].
Aging of blood vessels and progression of vascular pathology is observed in the presence of diabetes mellitus. Impaired glucose tolerance occurs in 50% of patients [9], the prevalence of type 2 diabetes is determined by the occurrence of the genetic variant: for XP deletion, the prevalence of diabetes is 9% relative to the general population, with 45.X - 18%, XP - 23%, isochromosome Xp - 43%. In this case, vascular sympathetic dysfunction develops due to the formation of diabetic autonomic neuropathy, estrogen deficiency and an imbalance in the renin-angigotensin-aldosterone system [6]. It is advisable to treat patients with Shereshevsky-Turner syndrome together with a cardiologist and endocrinologist.
Monitoring of the course of cardiac pathology is carried out according to indications, for arterial hypertension - annually. For patients with practically normal blood pressure and cardiovascular system, repeated imaging of the heart and aorta is carried out upon transfer to an adult clinic according to the patient’s age, before pregnancy, and with the initial detection of arterial hypertension. If cardiac sonography has already been performed, then according to age, MRI and further imaging every 5-10 years are indicated. If a cardiovascular disease is detected, monitoring and treatment by a cardiologist is indicated according to observation programs appropriate to the identified disease [7].
In patients with Shereshevsky-Turner syndrome, an atherogenic change in the lipid profile is observed: an increase in low-density lipoproteins and triglycerides, a decrease in high-density lipoproteins [13]. In the presence of cardiovascular disease, diabetes mellitus type 1 and 2 with target organ damage, chronic kidney disease with a glomerular filtration rate of less than 30 ml/min/1.73 m2 and a SCORE value of 10% or more, the patient should be considered very high risk. A high risk is established for familial dyslipidemia, severe arterial hypertension, diabetes mellitus without complications, a moderately reduced glomerular filtration rate of 30-59 ml/min/1.73 m2 and a SCORE value of 5% or more, but less than 10%. Moderate risk - with a SCORE value of 1% or more, but less than 5% over 10 years, as well as with a family history of cardiac pathology, abdominal obesity, low physical activity, changes in the values of cholesterol, triglycerides, C-reactive protein, established by high test systems sensitivity. Low risk is defined as a SCORE value of less than 1% and the absence of risk factors. Target values for the very high-risk group will be low-density lipoprotein levels of less than 1.8 mmol/L, high-risk - less than 2.5 mmol/L, moderate risk - less than 3 mmol/L. The target high-density lipoprotein value for women is a value of more than 1.2 mmol/L [14].
After laboratory confirmation of hypercholesterolemia, the percentage of cholesterol reduction in a particular patient to the target value should be determined depending on the risk subgroup. Next, the drug and dosage of the statin appropriate to the conditions are selected. Thus, to reduce low-density lipoproteins to 55%, rosuvastatin 20 mg or atorvastatin 80 mg per day should be used, and to reduce by 63%, only rosuvastatin at a dose of 40 mg per day should be used [15] (according to the FDA table - Food and Drug Administration, USA).
To achieve the target lipidogram level, statins should be prescribed at the highest recommended dose or the maximum tolerated by the patient. If you are intolerant to statins, it is recommended to use bile acid sequestrants or nicotinic acid preparations; under these conditions, it is possible to use cholesterol absorption inhibitors - alone or in combination with a bile acid sequestrant, or with a nicotinic acid preparation. If the target lipid profile is not achieved, a combination of a statin and a cholesterol absorption inhibitor or bile acid sequestrant or nicotinic acid may be recommended. Target triglyceride values of less than 1.7 mmol/l are achieved by administering fibrates [16].
When treating arterial hypertension, modern clinical guidelines should be used. Angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, calcium channel blockers are considered preferable for use; a combination of a beta-blocker and a thiazide diuretic seems convenient. Their combinations are possible, with the exception of the combination of the first two groups due to the established increased risk of cardiovascular events. If the risk is low, it is possible to use monotherapy with dose titration; if the risk is high, it is advisable to start treatment with combination drugs. In the future, their dosages are increased, including titration of two or more drugs until the maximum dose of each drug is reached. When resistant arterial hypertension is detected, 3-4 or more groups of drugs can be used when the maximum dose of each of these drugs is reached. The optimal combination is an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker in combination with a calcium channel blocker in maximum doses and a thiazide diuretic. Subsequently, the level of reninemia is tested. For low renin levels, aldosterone receptor blockers are additionally prescribed; for moderate increases, an alpha-adrenergic receptor blocker; for high levels, it is advisable to use a beta-blocker.
When analyzing electrocardiograms of patients with Shereshevsky-Turner syndrome, it was found that blockade of the posterior branch of the left bundle branch, accelerated atrioventricular conduction and T wave pathology are common, the PR interval is shortened and the QT interval is lengthened, a surrogate marker of repolarization heterogeneity, leading to a fatal outcome . In this regard, ECG testing is necessary when prescribing drugs that prolong the QT interval [17]. Appropriate criteria should be used to screen for probable long QT syndrome [18]. When carrying out genetic typing of patients with Shereshevsky-Turner syndrome, a connection between long QT and karyotype 45X was established; the SCN5A and KCNH2 genes (with large QT prolongation), as well as the KCNE2 gene (with small QT prolongation) [19]. To identify the risk group among blood relatives for sudden cardiac death in patients with long QT syndrome and for their dynamic monitoring, an adrenaline test can be used. When testing, you should use the current recommendations of the European Society of Cardiology for the diagnosis of ventricular arrhythmias [20]. All considered methods for searching for long QT syndrome should be widely used when counseling patients with Shereshevsky-Turner syndrome.
According to current clinical guidelines, long QT syndrome is diagnosed by repeating a 12-lead ECG with a QTc >480 ms or a score greater than 3 on the long QT syndrome risk scale. Regardless of QT duration, long QT syndrome is diagnosed by the presence of typical genetic mutations. The diagnosis of long QT syndrome is established by ECG when QTc >460 ms in a repeated 12-lead ECG in patients with unexplained syncopal episodes in the absence of secondary causes of QT prolongation [20].
For all patients with long QT syndrome, the following lifestyle modifications are recommended: 1) avoid drugs that prolong the QT interval (https:// www.crediblemeds.org); 2) correction of dyselectrolythemia (hypokalemia, hypomagnesemia, hypocalcemia) due to diarrhea, vomiting, metabolic disorders; 3) avoid triggers of genotypic arrhythmia (swimming with exercise, especially in LQTS1, and loud sounds in LQTS2). In drug treatment for clinically diagnosed long QT syndrome, beta-blockers are recommended for patients. For occasional mutations of long QT syndrome, the latter should be recommended in the presence of a normal QT interval. In cases of previous cardiac arrest while using beta-blockers, implantation of a cardioverter-defibrillator is recommended for patients with long QT syndrome. In addition to beta-blockers, implantation of a cardioverter-defibrillator should be recommended in patients with long QT syndrome who have experienced syncope and/or ventricular tachycardia while taking adequate doses of beta-blockers. Sympathetic denervation of the left departments should be recommended for symptomatic long QT syndrome: a) beta-blockers are ineffective, there is intolerance, they are contraindicated; b) therapy by implantation of a cardioverter-defibrillator is contraindicated or rejected; c) patients using beta-blockers with marked multiple shocks during therapy with an implanted cardioverter-defibrillator [20].
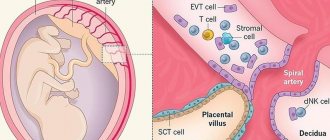
An important area of advisory support for patients with Shereshevsky-Turner syndrome is the period of observation during pregnancy and childbirth. Fertilization technology using donor eggs has been available for 20 years, while in the USA in 1997, out of 258 women in the assistance program, 52% received treatment for Shereshevsky-Turner syndrome [21]. Puberty may appear in 10% of patients with somatic signs of Shereshevsky-Turner syndrome, while spontaneous pregnancy is possible in 2% of women in this group [22]. Their pregnancy is accompanied by an increase in the risk of maternal mortality by 150 times relative to the general population of pregnant women [23].
When planning pregnancy, a patient with Shereshevsky-Turner syndrome is informed about the possibility of inheriting genetic abnormalities during spontaneous pregnancy, which requires participation in a consultation with a specialist in medical genetics. The possibility of influence on the course of pregnancy by cardiovascular (arterial hypertension, preeclampsia, aortic rupture) and metabolic complications (mellitus, gestational diabetes), possible complications when using medical technologies to support childbirth (caesarean section is performed in 85% of cases) is discussed. . Due to the development of obstetric or cardiovascular complications for the fetus, premature birth, intrauterine growth retardation and the need for intensive care related to the newborn are possible [24].
Screening before pregnancy includes: examination by a cardiologist for the presence of congenital heart disease with assessment of the degree of hemodynamic disorders; conducting ECG, echocardiography, MRI, control of arterial hypertension, examination of carbohydrate metabolism (fasting glycemia and HbA1c), abdominal ultrasound and examination of liver and kidney function, Papanicolaou test and ultrasound gynecological examination [25]. Pregnancy is contraindicated in the presence of a history of surgical treatment of the aorta, aortic dissection, dilatation of the aorta with a maximum size of more than 25 mm/m2 or 35 mm, coarctation of the aorta, uncontrolled arterial hypertension (despite treatment), as well as portal hypertension with dilatation of the esophageal veins [24].
Complications from the cardiovascular system during pregnancy include aortic dissection due to congenital defects and arterial hypertension. The identified progression factors that contribute to the manifestation of complications include an aortic size index of more than 2.0 cm/m2, cardiac anomalies - 25-50% of cases, arterial hypertension - 40-50%, karyotype 45.X. Arterial hypertension induced by pregnancy contributes to the development of preeclampsia and eclampsia with complications in the mother and fetus. With initially diagnosed arterial hypertension, preeclampsia, eclampsia, and HELLP syndrome are 50% more likely to develop [25]. Pregnancy is possible if the aorta has a diameter of less than 25 mm/m2 or 35 mm if there is no connection with coarctation of the aorta. In case of a verified increase in aortic dilatation by 10% or more, other imaging techniques (MRI, CT or transesophageal echocardiography) should be recommended. Pregnancy is contraindicated if documented aortic dilatation is detected [24].
Coordinated cardiovascular monitoring and echocardiography are indicated in the following periods: at the end of the first and second trimesters; monthly in the third trimester, MRI is recommended if the aortic diameter increases by 10% or more between two echocardiography studies. If aortic dilatation develops, hospitalization will be required in a clinic with a cardiac surgery department and a maternity hospital with a neonatology department and/or neonatological intensive care unit for gestational age more than 32 weeks, cardiological and surgical advisory assistance from a specialized center; monitoring the development of the fetal pulmonary system during childbirth at 25-34 weeks; planning a caesarean section. In the absence of dilatation, cesarean section is indicated in 85% of patients after 34 weeks due to an anatomically narrow pelvis, depending on the condition of the cardiovascular system. Natural childbirth occurs with blood pressure monitoring, in the absence of fetal/pelvic size disproportion. It is recommended to use a birth aid (vacuum extraction or the application of surgical forceps). Childbirth is carried out with the participation of a team of specialists, including a cardiologist and a cardiac surgeon [24].
The risk/benefit ratio should be assessed when prescribing the following drugs during pregnancy: amiodarone, clopidogrel, IIb/IIIa receptor inhibitors, prasugrel hydrochloride, propafenone hydrochloride, ticagrelor, warfarin. Drugs: adenosine, acetylsalicylic acid, beta-blockers (excluding atenolol), hydralazine, methyldopa, nitrates, nifedipine, sotalol, digoxin, diltiazem, verapamil, flecainide, furosemide, unfractionated heparin - assessed as safe for use during pregnancy [26]. Low molecular weight heparins are used in the treatment program for pulmonary embolism with existing coagulopathy (enoxaparin 1 mg/kg every 12 hours or deltaparin 100 IU/kg every 12 hours; maximum level of anti-Xa activity 4-6 hours after drug administration is 0.6–1.2 IU/ml) [27]. When using drugs that prolong the QT interval, the risks and benefits of their use in Shereshevsky-Turner syndrome should be further assessed.
In the postnatal period, cardiovascular risk remains, as a result of which the size of the aortic root should be monitored according to the examination protocol on the 5th and 8th day after birth in a specialized sonographic center. In the prenatal period, it is possible to examine the fetus by sonography and karyotyping of chorionic villi cells or amniotic fluid. Testing a newborn for the presence of Shereshevsky-Turner syndrome is performed during spontaneous pregnancy [24]. In the postnatal period, it is possible to carry out cell karyotyping, PCR diagnostics, and search for Y-mosaicism [28].
Modern clinical cardiology practice is constantly being improved through the development and application of clinical guidelines for the management of patients with arterial hypertension, coronary heart disease, and heart defects. The presented materials, developed by foreign experts, on advisory cardiac care for patients with Shereshevsky-Turner syndrome contribute to the formation of a reasonable clinical use of diagnostic and therapeutic technologies for this genetically determined pathology, which is difficult to observe clinically.
Contact Information:
Kazakov Sergey Alekseevich - Ph.D., Associate Professor of the Department of Cardiology and Rheumatology.
Belarusian Medical Academy of Postgraduate Education.
220013, Minsk, st. P. Brovki, 3, bldg. 3; sl. tel. 375 17 331-92-82.
There is no conflict of interest.
Key words:
arterial hypertension, aortic dissection, coronary heart disease, long QT syndrome, Shereshevsky-Turner syndrome
Author(s):
Kazakov S. A.
Medical institution: Belarusian Medical Academy of Postgraduate Education
Turner-Shereshevsky syndrome: causes of infertility in patients
If the pathology was not identified in infancy, then with a high probability the diagnosis will be made during puberty. The reason for contacting a doctor will be a noticeable delay in the girl’s sexual development. An examination may show abnormal formation of the genital organs:
- funnel-shaped entrance to the vagina;
- underdevelopment of the labia minora, hymen and clitoris;
- high crotch;
- the appearance of the labia majora is more similar to the scrotum;
- pathological skin atrophy.
Moreover, when there is a suspicion of Shereshevsky-Turner syndrome, diagnostics may show that the girl’s uterus is undeveloped, instead of the ovaries there are bilateral strands of fibrous stroma, and the eggs do not mature in them.
Secondary sexual characteristics in such patients are weakly expressed. They do not have hair growth in the armpits and pubic area, and the mammary glands do not form properly. Most sick girls suffer from lack of menstruation (amenorrhea).
Obviously, with such a pathology there will be difficulties with procreation. Only five percent of patients are fertile and give birth to children without medical intervention. More often, if a diagnosis of Shereshevsky-Turner syndrome is made, infertility treatment is inevitable.
List of sources
- Dedov I.I., Peterkova V.A., Semicheva T.V., Volevodz N.N. Shereshevsky-Turner syndrome. - M., 2002;
- Kravets E.B. Clinical lectures on pediatric endocrinology. - Tomsk, Ex Libris, 2007;
- Peterkova V.A., Bezlepkina O.B., Semicheva T.V., Koledova E.B. Shereshevsky-Turner syndrome in children (clinic, diagnosis, treatment). Guidelines. M., 1998;
- Dedov I.I., Peterkova V.A. Pediatric endocrinology. Guide to pediatric endocrinology. - M., Universum Publishing, 2006;
- Pankratova, M.S. Shereshevsky-Turner syndrome in the practice of a pediatric endocrinologist / Pankratova M.S., Peterkova V.A. //Pediatrics. - 2009. - No. 4. - P. 115-121.
Paronychia
Paronychia is a purulent inflammation of the periungual fold and tissues at the base and sides of the nail.
The disease may be caused by Shereshevsky-Turner syndrome. Another cause of paronychia is infection under the skin as a result of injury, prolonged exposure to chemicals, or poor personal hygiene.
Basic information
A clear connection between the occurrence of Shereshevsky-Turner syndrome and age and any diseases of the parents has not been identified. However, pregnancies are usually complicated by toxicosis, the threat of miscarriage, and childbirth is often premature and pathological. Features of pregnancies and childbirth ending in the birth of a child with Shereshevsky-Turner syndrome are a consequence of chromosomal pathology of the fetus. Impaired formation of the gonads in Shereshevsky-Turner syndrome is caused by the absence or structural defects of one sex chromosome (X chromosome).
In the embryo, primary germ cells are formed in almost normal numbers, but in the second half of pregnancy they undergo rapid involution (reverse development), and by the time the child is born, the number of follicles in the ovary is sharply reduced compared to the norm or they are completely absent. This leads to severe deficiency of female sex hormones, sexual underdevelopment, and in most patients to primary amenorrhea (absence of menstruation) and infertility. The resulting chromosomal abnormalities are the cause of developmental defects. It is also possible that concomitant autosomal mutations play a certain role in the appearance of developmental defects, since there are conditions similar to Shereshevsky-Turner syndrome, but without visible chromosomal pathology and sexual underdevelopment.
Karyotype 45,(X0)=70% / 46,(XX)=30% - mosaic form of Turner syndrome.
In Shereshevsky-Turner syndrome, the gonads are usually undifferentiated connective tissue cords that do not contain gonadal elements. Less common are rudiments of the ovaries and elements of the testicles, as well as rudiments of the vas deferens. Other pathological findings are consistent with the clinical presentation. The most important changes in the osteoarticular system are shortening of the metacarpal and metatarsal bones, aplasia (absence) of the phalanges of the fingers, deformation of the wrist joint, and osteoporosis of the vertebrae. Radiologically, in Shereshevsky-Turner syndrome, the sella turcica and the bones of the cranial vault are usually not changed. Defects of the heart and large vessels are noted (coarctation of the aorta, patent ductus botallus, patent interventricular septum, narrowing of the aortic mouth), and renal malformations. Recessive genes for color blindness and other diseases appear.
Shereshevsky-Turner syndrome is much less common than trisomy X, Klinefelter syndrome (XXY, XXXY), and XYY, which indicates the presence of strong selection against gametes that do not contain sex chromosomes, or against XO zygotes. This assumption is confirmed by the fairly frequently observed monosomy X among spontaneously aborted fetuses. In this regard, it is assumed that surviving XO zygotes are the result not of meiotic, but of mitotic nondisjunction, or the loss of the X chromosome in the early stages of development. YO monosomy has not been detected in humans. Population frequency 1:1500.
All regions of the world and culture are affected by this pathology approximately equally. It is estimated to occur in 3% of all human fetuses. However, only 1% of these fetuses survive birth.
Causes
The likelihood of developing the syndrome increases if there were genetic diseases in previous generations. There are a number of other unfavorable factors leading to mutations in genetic information. Among them are chronic alcoholism of the father and mother, and the unfavorable state of the environment in the region.
No relationship was found between the age of the parents and the development of the disease. However, it has been noted that pregnancies with such fetal pathologies usually occur with toxicosis, there is a threat of miscarriage, and the child is born prematurely. The nature of these features is in the chromosomal pathology of the fetus.
Treatment
At the first stage, therapy consists of stimulating body growth with anabolic steroids and other anabolic drugs. Treatment should be carried out with minimal effective doses of anabolic steroids intermittently with regular gynecological monitoring. The main type of therapy for patients is estrogenization (prescription of female sex hormones), which should be carried out from 14-16 years of age. Treatment leads to feminization of the physique, the development of female secondary sexual characteristics, improves trophism (nutrition) of the genital tract, and reduces the increased activity of the hypothalamic-pituitary system. Treatment should be carried out throughout the entire childbearing age of patients.
If, with the help of hormonal therapy, it is possible to grow the uterus to a normal size, then pregnancy in such patients is possible using IVF with a donor egg. Cases where eggs have been preserved are rare.
Recently, growth hormone therapy has been used to increase final growth rates.