- How common are neuroblastomas?
- Risk factors for neuroblastoma
- What are the causes of neuroblastoma?
- Is it possible to prevent the development of neuroblastoma?
- Is early diagnosis of neuroblastoma possible?
- How is neuroblastoma diagnosed?
- Treatment of neuroblastoma in children
- What happens after treatment for neuroblastoma ends?
Neuroblastoma is a type of malignant tumor and usually occurs in infants and children and very rarely in children over 10 years of age. The cells of this tumor resemble nerve cells in the early stages of their development in the fetus.
One third of neuroblastomas develop in the adrenal glands, the other third in the abdominal cavity along the nerve trunks along the spine, and the rest in the chest cavity and neck. Some neuroblastomas arise from the spinal cord. Sometimes, due to the wide distribution of the tumor at the time of diagnosis, it is difficult to determine the exact location where the tumor originated.
Not all tumors of the nervous system are malignant.
Ganglioneuroma is an example of a benign tumor.
Ganglioneuroblastoma is a mixed tumor and contains both malignant and benign areas. Immature (malignant) cells from this tumor can grow and metastasize.
Ganglioneuromas are usually removed surgically and then carefully examined under a microscope to determine whether there are areas of ganglioneuroblastoma. If ganglioneuroma is diagnosed, no additional treatment is required. In contrast, ganglioneuroblastomas are treated in the same way as neuroblastomas.
Neuroblastoma is an unusual tumor for many reasons. This tumor secretes a hormone that leads to unusual changes in the body, for example, rotational movements of the eyeballs, spastic muscle twitching, and the presence of constant loose stools.
These changes are called paraneoplastic syndromes.
Neuroblastoma can behave in unusual ways. Sometimes tumor cells spontaneously die and the tumor disappears. This phenomenon occurs most often in very young children and much less frequently in older patients. In some cases, tumor cells spontaneously mature and stop dividing. Thus, neuroblastoma turns into ganglioneuroma, a benign tumor.
Causes and risks
The reasons for the appearance of the tumor are not fully understood. Neuroblastoma occurs when normal neuroblasts do not mature into nerve cells. Instead, they continue to grow and divide uncontrollably. It is believed that these immature (embryonic) nerve cells begin to mutate even before the birth of a child, when chromosomes begin to change and/or a failure in the regulation of gene function occurs. As current research confirms, in most children the disease is not hereditary. Only about 1% of neuroblastoma cases are hereditary, most often due to mutations in a gene called ALK (or anaplastic lymphoma kinase gene) or in the PHOX2B gene. To date, there are no clinical studies proving that external factors (environment, harmful working conditions of the child’s parents, taking any medications, smoking and drinking alcohol during pregnancy) can cause the disease.
Clinical picture of neuroblastoma
Neuroblastoma can occur in any anatomical region where the sympathetic nervous system is located: adrenal glands, cervical, thoracic and abdominal sympathetic divisions, paraganglia.
The most common location of neuroblastoma is the retroperitoneal space (the tumor is equally often diagnosed in the adrenal glands and paravertebral retroperitoneal ganglia). In 15% of children, neuroblastoma is localized in the posterior mediastinum. Less commonly, tumors occur in the pelvis (6%) and neck (2%) (Fig. 1).
Rice. 1. Localization of neuroblastoma
In the early stages of the disease, many children may not have any symptoms. Their tumor is discovered by chance, for example, during a routine ultrasound examination of infants during medical examination. As a rule, complaints in children appear when the tumor has already grown significantly and puts pressure on neighboring organs, disrupting their function, or when the tumor has metastasized.
Common symptoms of the disease include the following: weight loss, weakness, bone and joint pain, and intractable diarrhea.
Most manifestations of the disease and their severity depend on the location of the tumor and its relationship with neighboring organs and tissues. Thus, patients under 2 years of age with retroperitoneal localization are characterized by an increase in abdominal size, fever and weight loss. Neuroblastoma of the retroperitoneal space is palpated through the anterior abdominal wall in the form of a tuberous, non-displaceable tumor node (Fig. 2). In older children, the disease can manifest as pain caused by metastatic bone lesions, respiratory disorders, an increase in the size of the abdomen, and the appearance of constipation. In patients with large retroperitoneal tumors, a developed network of saphenous veins and expanded edges of the chest can be found.
Rice. 2. Neuroblastoma of the retroperitoneum
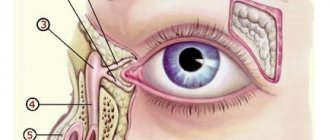
Localization of the tumor in the cervicothoracic region of the sympathetic spine causes Horner's syndrome (constriction of the pupil, different pupil sizes, redness and/or drooping of the upper eyelid and slight elevation of the lower). Other changes in the eye area may include hemorrhages in the skin or mucous membrane, “bruising” on the eyelids and under the eyes. In the later stages of the disease, black circles sometimes appear around the eyes (spectacle-shaped hematoma) (Fig. 3).
Rice. 3.1. Symptom of "glasses"
Rice. 3.2. Symptom of “glasses” with metastases in the orbit
Damage to the posterior mediastinum can cause a dry cough, respiratory distress, chest deformation, and frequent regurgitation. When the tumor is localized in the pelvic cavity, dysfunction of the pelvic organs (periodic urinary incontinence, defecation disturbance), and edema of the lower extremities are noted.
When the tumor spreads into the spinal canal and compresses the spinal cord, weakness in the legs, unsteady gait, paralysis of the lower extremities, as well as dysfunction of the pelvic organs (urinary retention or involuntary urination, constipation) may develop.
Rarely, in approximately 2-4% of all cases, children have a cerebellar syndrome associated with neuroblastoma (“opsoclonus-myoclonus” or Kinsburne encephalopathy), characterized by uncoordinated, irregular movements of the trunk and limbs, myoclonus and chaotic movements of the eyeballs.
Neuroblastoma is a hormone-producing tumor capable of secreting physiologically active substances - catecholamines - adrenaline, norepinephrine and dopamine. In urine, as a rule, the excretion of their metabolites – vanillylmandelic and homovanillic acids – is increased. In 95% of cases, the higher the degree of malignancy, the greater the hormonal activity of neuroblastoma. The effects of secreted hormones cause specific clinical symptoms of neuroblastoma - a sharp increase in blood pressure, diarrhea (caused by the secretion of vasoactive intestinal polypeptide), sweating, emotional lability, periodic fever.
Neuroblastoma metastasizes most often to the bone marrow, bones, distant lymph nodes, liver or skin, and rarely to the brain or lungs. Signs of distant metastases can be detected in the form of exophthalmos, hemorrhages in the orbits, and tumor nodes on the head.
Among the features of the course of neuroblastoma are the following:
- Regression is the ability of a tumor to spontaneously or induced by minimal chemotherapy or radiation therapy to decrease (involution). As a rule, this occurs in children under 1 year of age with stage IVS of the disease.
- Reversion is the ability of a tumor to transform from more malignant forms into benign ganglioneuromas (spontaneously or induced).
- In some cases, the tumor process has the opposite direction: a tendency towards an aggressive course and rapid metastasis.
Forecast
The survival prognosis for neuroblastoma is determined by a number of factors: the age of the child, the stage of the disease, and the morphohistological structure of the tumor. Accordingly, with modern neuroblastoma treatment programs, the overall 3-year survival rate of patients is:
- at stages 1 and 2 of the disease varies between 95-99%,
- at stage 3 this figure is 80-98%;
- survival rate for stage 4 ranges from 60-76%.
At the same time, the prognosis is more favorable for children under 1 year of age. Regarding tumor location, the maximum number of unfavorable outcomes is observed when neuroblastoma is localized in the retroperitoneal space, and the minimum number is observed when localized in the mediastinum.
Diagnosis of neuroblastoma
Laboratory methods
In a clinical blood test, if the bone marrow is damaged, a decrease in blood parameters may be observed: anemia, leukoneutropenia, thrombocytopenia.
Neuroblastoma-specific markers can also be determined in the blood serum: neuron-specific enolase (NSE), levels of catecholamine metabolites, lactate dehydrogenase (LDH) and ferritin.
An easy-to-perform specialized test for diagnosing neuroblastoma is to determine the level of vanillylmandelic and homovanillic acids in a urine test. They are also called the “urinary catecholamine metabolite test.” This analysis can be carried out by collecting the child’s urine over the course of a day or in a single portion of urine.
Before starting specific therapy, the diagnosis is always verified; The diagnosis of neuroblastoma is made by histological examination of a biopsy of the primary tumor or metastases, or by a combination of the presence of tumor cells in the bone marrow and an increased level of daily excretion of catecholamines.
Molecular genetic research
To determine the degree of malignancy of neuroblastoma, a number of genetic studies are performed: MYCN mutation, deletion of chromosomes 1p or 11q. The presence of these mutations is an unfavorable prognosis factor for the disease.
Instrumental methods
Ultrasonography
The ultrasound method of examination allows us to detect the presence of a tumor and its location in relation to the internal organs. Ultrasound allows you to assess the degree of local and distant spread of the malignant process and reveals metastatic damage to the liver and lymph nodes (Fig. 4).
Rice. 4. Ultrasound of the formation of the right adrenal gland
Computed tomography of the abdominal cavity and retroperitoneum
Computed tomography provides more detailed information about the anatomical relationship of the tumor to surrounding tissues and organs and allows one to assess the structure of the tumor (Fig. 5).
Rice. 5. CT scan of a neurogenic formation in the retroperitoneal space on the right
Magnetic resonance imaging of the abdominal organs, retroperitoneum and spine
MRI of the abdominal cavity and retroperitoneal space is a safe and highly informative research method that allows not only to clarify the location of the tumor, the exact size of the tumor and its relationship with surrounding tissues, but also to identify metastatic foci in the liver and abdominal cavity. MRI also makes it possible to assess the condition of the bone structures and soft tissues of the spinal column and spinal canal (Fig. 6).
Rice. 6. MRI of a neurogenic formation in the retroperitoneal space on the left
Radioisotope research
One of the most informative specific methods for topical diagnosis of neuroblastomas is scintigraphy with metaiodobenzylguanidine (MYBG) labeled with 123I. MyBG is similar in chemical structure to catecholamines in the body. This substance is concentrated in those tumors that produce catecholamines. MYBG is labeled with a radioactive isotope of iodine (for example, 123I) in a dose that is safe for life, and the finished drug is injected intravenously into the bloodstream. Accumulating in tumor cells, it emits radiation. These signals are recorded by a special camera and translated into an image. Thus, the method makes it possible to detect not only the primary tumor, but also the presence of regional and distant metastases (Fig. 7).
Rice. 7. Scintigraphy with metaiodobenzylguanidine (visualizes a primary paravertebral tumor with metastases to the skull bones)
Bone scintigraphy is a method of scanning skeletal bones using intravenous administration of a radiopharmaceutical. The radiotracer accumulates in areas affected by tumor cells in the bones, as seen in gamma camera images. Thus, this research method allows us to identify possible metastases of skeletal bones.
Bone marrow examination (myelogram and trephine biopsy) is used to determine the presence of tumor cells in the bone marrow.
Classification
There are different classifications of neuroblastoma based on tumor size/extension, one of which is summarized below:
- The first stage is characterized by the presence of a single node not exceeding 5 cm with the absence of metastases.
- The second stage is characterized by the presence of a single node, the size of which can vary between 5-10 cm, metastases , and the lymph nodes are not affected.
- The third stage - the size of the node does not exceed 10 cm, damage to the lymph nodes is noted, there are no metastases to distant organs, or the size of the tumor exceeds 10 cm without metastases .
- Fourth (A) stage - a tumor of any size is determined, lymph nodes are involved in the process and the presence of distant metastases.
- Fourth (B) stage – multiple tumor nodes with synchronous growth or a large single neoplasm, metastases to adjacent and distant organs are determined.
Treatment of neuroblastoma
Taking into account various scenarios for the development of disease outcome (from spontaneous regression to death from progression), risk-adapted principles are proposed in the treatment of neuroblastoma. Patients are stratified according to risk factors before treatment (usually clinical and molecular genetic data) and based on tumor response during treatment (response-adapted). When identifying risk groups before starting treatment, the following prognostic criteria are used: stage of the disease, age at diagnosis, results of molecular genetic data - MYCN amplification and deletion of the short arm of 1p (Fig. 8).
Rice. 8. Stratification of patients into risk groups according to the presence of unfavorable prognosis factors
Neoadjuvant (preoperative) polychemotherapy
- The need for polychemotherapy is determined by the stage of the disease and the presence or absence of unfavorable clinical and biological factors (n-myc amplification, short arm 1p deletions).
- Polychemotherapy allows you to reduce the size of the primary tumor to make surgery safer and more radical.
- The basic drugs for the treatment of neuroblastoma are vincristine, cyclophosphamide, anthracyclines, ifosfamide, platinum drugs, etoposide.
Features of surgical treatment
- For localized stages of the disease, treatment may be limited to complete removal of the tumor.
- In stages III-IV of the disease, the surgical stage is carried out after preoperative (neoadjuvant) polychemotherapy, which is aimed at reducing the size of the tumor with the aim of subsequent radical removal of the tumor.
- In cases where complete tumor removal is impossible, treatment is supplemented with radiation therapy in the postoperative period.
Adjuvant (postoperative) polychemotherapy
Drug treatment should be started 5-7 days after surgery. In the absence of indications for 2nd line polychemotherapy, treatment regimens use the same basic drugs as for neoadjuvant therapy.
Radiation therapy
- Radiation therapy is used to destroy any tumor cells that cannot be removed by surgery and/or that were not destroyed by previous therapies.
- If the operation is non-radical and involves the presence of an active residual tumor, the residual tumor tissue, the lymph nodes involved in the process, and the areas of communication of the tumor with surrounding organs and tissues are irradiated.
- Irradiation is carried out daily, starting from the 5-7th day after surgery.
- According to indications, irradiation of metastatic foci is carried out (for therapeutic, sometimes for analgesic purposes).
If there are unfavorable prognostic factors, in particular the detection of a MYCN oncogene mutation in the tumor, high-dose chemotherapy with autologous hematopoietic stem cell transplantation is recommended.
For patients with a high risk of relapse (advanced stages of the disease with the presence of unfavorable molecular genetic markers), “maintenance” differential therapy with 13-cis-retinoic acid and passive immunotherapy with anti-GD2 monoclonal antibodies is recommended.
Immunotherapy
Immunotherapy is a new treatment for neuroblastoma that is used to destroy cancer cells. Disialoganglioside GD2 is an antigen molecule expressed by neuroblastoma cells, making it an ideal target for antibody immunotherapy: the antibody acts against this molecule. Immune system cells may contribute to the death of neuroblastoma cells.
At the National Medical Research Center of Oncology named after. N.N. Petrov performs all stages of treatment, including minimally invasive operations (laparoscopic and thoracoscopic), tandem transplantation and passive immunotherapy.
By intensifying therapy in children with high-risk neuroblastoma (with the inclusion of tandem high-dose polychemotherapy in consolidation and passive immunotherapy in post-consolidation), it was possible to increase survival results by 20%.
A comment
The presented work is a description of clinical observations of pediatric patients with neuroblastoma and signs of involvement in the process of the central nervous system - the brain and spinal cord. Based on the examples given, the authors analyze the current state of the problem from the point of view of the feasibility of neurosurgical interventions.
The modern approach to the treatment of neuroblastoma (a highly malignant embryonal tumor) reflects the general trends in oncology: obtaining a histological diagnosis, molecular genetic typing, clinical staging and “standard” combination treatment. In most cases, the issue of surgical treatment is a matter of the second or third stage. Historically, patients with neuroblastoma and signs of spinal cord compression were treated by a neurosurgeon as the first step—urgent decompression of the spinal cord by removing the tumor from the spinal canal. The most significant negative event of this tactic is the relatively high incidence of spinal deformities in the long-term period. Modern series of patients have shown that in some cases, chemotherapy in the form of “neoadjuvant” treatment also achieves good results in terms of spinal cord decompression due to the rapid reduction of tumor volume. It is this idea that the authors convincingly illustrate with their clinical examples.
In my opinion, the work is valuable for two reasons. First: the authors are the first in Russian neurosurgical literature to raise the question of the inappropriateness of mandatory removal of primary neuroblastoma from the spinal canal. Second, perhaps more important: the authors show that any “standard tactics” of treatment in real clinical conditions is not absolute and, despite advances in oncology, patients with rapidly increasing reversible neurological deficits remain candidates for urgent neurosurgical interventions.
Yu.V. Kushel (Moscow)
Recommendations after treatment
- After successful treatment of neuroblastoma, the child must undergo medical supervision, which may include examination and consultation with a pediatric oncologist, a blood test for tumor markers (LDH, NSE), a urine test for catecholamine metabolites, ultrasound and CT/MRI, scintigraphy with MYBG and other studies.
- The pediatric oncologist conducts an examination once every 1.5-2 months during the first year after the end of treatment, once every 3 months during the 2nd year, once every 6 months over the next 2 years, then 1 once a year.
- Patients are not removed from the register.
BIBLIOGRAPHY
- Weiner M.A., Queiro M.S. Secrets of pediatric oncology and hematology. – M.: Binom, Dialect, 2008.
- Pediatric oncology. National leadership / Ed. M. Aliyev, V. Polyakov, G. Mentkevich, S. Mayakova. – M.: Practical Medicine, 2012.
- Pediatric Oncology: A Guide for Doctors / Ed. M.B. Belogurova. – St. Petersburg: SpetsLit, 2002.
- Durnov L.A., Goldobenko G.V. Pediatric oncology: Textbook. – 2nd ed. Reworked and additional – M.: Medicine, 2002.
- Imyanitov E.N., Hanson K.P. Molecular oncology - St. Petersburg: St. Petersburg MAPO, 2004.
- Clinical lectures on pediatric oncology. In 2 parts / Under. ed. L.A. Durnova. – M.: MIA, 2006.
- Clinical recommendations. Oncology / Ed. IN AND. Chissova, S.L. Daryalova. – M. GEOTAR-Media, 2008.
- Clinical lectures on pediatric oncology vol. 1 /ed. L.A. Durnova, Moscow, 2004.
- Clinical lectures on pediatric oncology vol. 2 /ed. L.A. Durnova, Moscow, 2006.
- Kozlova S.I., Demikova N.S., Semanova E.M., Blinnikova O.E. Hereditary syndromes and medical genetic counseling - M., Praktika, 1996.
- Guide to pediatric oncology / ed. acad. RAMS L.A. Durnova, M. Ed. "MIKLOSH", 2003
- Neuroblastoma. Information for parents. - Moscow, 2021. - 96 p./ Goleva M.N., Emelian E.N., Zakharova A.N., Kovalenko Yu.S., Ruksova O.A.
- Neuroblastoma UK / Children's Cancer and Leukaemia Group (CCLG), 2015
- Nai-Kong V. Chenng Neuroblastoma, 2005.
- Pizzo Ph.A., Poplack DG Principles and Practice of Pediatric Oncology, 6th Edition, 2011.
Authors:
- Izmozherova Rina Igorevna;
- Senchurov Evgeny Mikhailovich;
- Mikhailova Elena Andreevna;
- Ivanova Svetlana Vyacheslavovna;
- Faseeva Natalya Dmitrievna;
- Borokshinova Ksenia Mikhailovna;
- Zhuk Irina Nikolaevna.