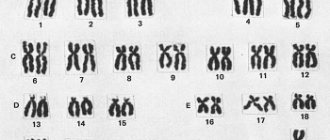
A person's genetic sex depends on the presence or absence of a Y chromosome. The cells of a female body have two sex X chromosomes, while the cells of a male body have one X and one Y chromosome. Genetic sex determines the true sex, which is associated with the structure of the gonads. Primary sexual characteristics, i.e. the genital organs, which include the gonads, begin to form in accordance with the hereditary information in the embryo at the 4th week of development. At earlier stages, the rudiments of the genital organs are the same in both the male and female organisms.
Diseases of the male reproductive glands
The male gonads or testes are a paired organ.
They produce male sex hormones - androgens, the main of which is testosterone, since it has the greatest biological activity. Androgens are involved in the development of secondary male sexual characteristics, skeletal formation and spermatogenesis. They are responsible for sexual function, their quantity has a direct impact on potency and libido. In addition, testosterone has pronounced anabolic activity, it stimulates protein synthesis and is involved in maintaining the constancy of the protein composition of the blood. The secretory activity of the testes is under the control of gonadotropic hormones produced by the anterior pituitary gland.
Thyroid hormones
Thyroid hormones are components that are synthesized by the thyroid gland. They take part in various physiological processes of the body. They contain an iodine component.
Thyroid hormones are:
- Free thyroxine, triiodothyronine.
- Total thyroxine, triiodothyronine.
- Thyroxine-binding globulin and thyroglobulin.
The thyroid gland also produces calcitonin and parathyroid hormone, these substances control the process of calcium metabolism.
Thyroid hormones stimulate growth and development of the body, affect the heartbeat, and help remove excess fluid from the body. They affect physical and mental work, and also stimulate metabolism.
The ovaries contain receptors that respond to thyroid hormones. Thanks to them, the growth of follicles is stimulated, the production of progesterone and estrogens is enhanced.
These substances help reduce the production of glycogen and fat deposits. A large amount of thyroid hormones slows down protein metabolism, and a low concentration of these substances promotes protein synthesis. A thyroid hormone test can determine the presence of thyroid disease.
The most common diseases of the male gonads are:
- Hypogonadism is a pathological condition of the body characterized by insufficient production of male sex hormones. It manifests itself in disruption of the processes of spermatogenesis, underdevelopment of the external and internal male genital organs and secondary sexual characteristics.
- hyperprolactinemia is an increased level of the hormone prolactin in the blood; it is caused by hypothalamic-pituitary dysfunction.
- male menopause - insufficiency of the function of the male gonads in adulthood and old age.
The most common hormone-dependent ovarian diseases include:
- hypogonadism is a pathological condition manifested in the production of insufficient amounts of female sex hormones by the ovaries.
- amenorrhea with normally developed sexual characteristics - absence of menstruation due to a lack or excess of gonadotropic hormones - FSH and LH.
- amenorrhea with androgen excess
- hirsutism is a male type of skin hair growth in women. It manifests itself in the growth of hair on the face, chest, abdomen, back, around the nipples of the mammary glands and on the thighs.
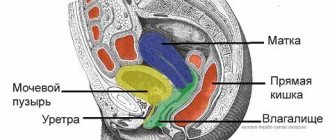
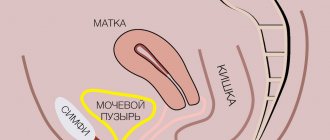
The most common symptom of abnormalities in the structure or functioning of the ovaries is menstrual irregularities. They manifest themselves in the complete absence of discharge - amenorrhea, a change in their quantity, and in different shifts in the timing of the next menstruation. Amenorrhea is not an independent disease, but a symptom of a disease affecting the organs of the reproductive and endocrine system. To effectively treat such conditions, a thorough diagnosis is necessary, the purpose of which is to establish the true cause of the disease. This requires consultation with doctors of two specialties - a gynecologist and an endocrinologist.
Publications in the media
Diagnosis and clinical problems associated with distorted sexual differentiation require an understanding of the mechanisms of development of the gonads and ducts.
Normal sexual differentiation . The gonad primordia in humans do not develop sexual differentiation until the 45th–50th day of intrauterine development. In the early stages, internal genital ducts are present: both male (Wolffian, or mesonephric duct) and female (Müllerian, or paramesonephric duct) and undifferentiated gonads (genital ridges). Later (critical phase - 8 weeks of intrauterine development), differentiation of male and female genital organs occurs in various ways dictated by genetic and hormonal factors.
• Differentiation according to the male type. Genotype - 46,XY • Y-chrom. - male sex determinant. Under the influence of a regulatory factor (a factor that determines the development of testicles), encoded by the Y chromosome, the genital ridges develop as testicles; in the absence of the factor, the ovaries develop • Leidig cells of the fetal testicles, under the control of gonadotropins (chorionic and pituitary), secrete testosterone •• Under the influence of testosterone, the following develop from the mesonephric duct: vas deferens, epididymis, seminal vesicles •• 5-Reductase catalyzes the conversion of testosterone to dihydrotestosterone ( DHT), necessary for differentiation of the external genitalia (scrotum, penis), which is completed by 12–14 weeks of intrauterine development •• Sertoli cells of the fetal testicles secrete Müllerian inhibitory factor (MIF), which causes regression of the Müllerian ducts in the male fetus.
• Differentiation according to the female type (genotype - 46,XX) occurs in the absence of the factor Y-chromium, testosterone, DHT and MIF, which determines the development of the testicles. • In the absence of Y-chrom. genital ridges develop as ovaries • In the absence of MIF, the Mullerian duct develops into the fallopian tubes, uterus and upper third of the vagina • In the absence of testosterone and DHT, the Wolffian duct degenerates and the clitoris, labia majora and minora, and vagina develop • The ovaries begin to function at puberty; formation according to the female phenotype occurs autonomously, under the influence of hormones of the placenta and the maternal body. Distortions of sexual differentiation lead to the birth of a child who has characteristics of both male and female, but is not completely (phenotypically!) either male or female.
• True hermaphroditism . The gonads contain tissue from both the testes and ovaries. Karyotype: in approximately 80% - 46.XX, the remaining cases - 46.XY or mosaicism. The etiology is unclear •• There is usually significant virilization, as a result of which most true hermaphrodites are raised as males. Gynecomastia and cyclic hematuria may occur as a result of uterine bleeding. • A serious suspicion of true hermaphroditism should arise if the child has transitional type genitalia, an XX karyotype and a normal level of 17-hydroxyprogesterone, which excludes 21-hydroxylase deficiency. The final diagnosis is based on surgical exploration and detection of the gonads containing tissue from both the ovary and testes.
• Mixed dysgenesis of the gonads is observed with karyotype 45,X/46,XY •• Clinic: a wide range of structure of the external genital organs - from completely male to completely female ••• The gonads can look different: from externally recognizable ovaries to dysgenetic testes; Gonadal asymmetry is often observed ••• Effects of the 45,X cell line may mimic the phenotype of Turner syndrome •• Diagnosis is made by karyotyping.
• Male pseudohermaphroditism . Children of genotype 46,XY; there are testicles, but masculinization is incomplete (hypospadias, microphallia, underdeveloped scrotum with or without testicles). Male pseudohermaphroditism is observed in a variety of endocrine disorders (defects in testosterone synthesis, its metabolism and effects on target cells) •• Reifenstein syndrome. Familial form of male pseudohermaphroditism: phenotypically indeterminate gender of the genitals, hypospadias, postpubertal gynecomastia; infertility due to sclerosis of the seminiferous tubules. Synonym: Klinefelter–Reifenstein–Albright syndrome •• Testicular feminization (see Testicular feminization syndrome) •• Male pseudohermaphroditism (*300018, Xp21.3, DSS gene, À ) is a locus on the X chromosome, the duplication of which leads to sex inversion.
• 5a-reductase deficiency (*264600, r) impairs the conversion of testosterone to DHT •• Boys (karyotype 46XY) are born with external genitalia of indeterminate gender. Some newborns are classified as girls due to minimal evidence of virilization at birth •• Testosterone-dependent changes occur during puberty: movement of the testes to their final position, increase in muscle mass, deepening of the voice •• There is a report of one isolate in which many children were raised as girls, but after puberty they acquired pronounced features of a male phenotype •• Diagnosis can be made by detecting an increased testosterone/DHT ratio after HHT stimulation.
• Disorders of testosterone synthesis and metabolism are rarely observed; Several forms of enzyme deficiency are known (r) •• Cholesterol desmolase deficiency. A severe form of salt loss is characteristic. Severe deficiency of mineralocorticoids and GCs, as well as androgens, leads to death in early childhood, despite steroid hormone replacement therapy •• 3b-hydroxysteroid dehydrogenase deficiency. Men are not completely virilized (defect in testosterone synthesis). Women can be mildly virilized. The diagnosis is based on the detection of elevated blood concentrations of dehydroepiandrosterone and 17-hydroxypregnenolone •• 17a-hydroxylase deficiency •• 17-oxidoreductase deficiency prevents the conversion of androstenedione to testosterone. The diagnosis can be established by detecting an increased ratio of androstenedione to testosterone after HCG stimulation •• Testicular 17-a-ketosteroid reductase deficiency (*264300, 9q22, HSD17B3 gene, EDH17B3, r) - clinically indistinguishable from 5-a-reductase-2 deficiency (264600) •• 17,20-desmolase deficiency (*309150, À r) is a rare cause of male pseudohermaphroditism resulting from a block in the conversion of progestogens to androgens. The defect can be detected by a distorted ratio of progestogens and androgens both at basal levels and after stimulation with hCG. In women, the genitals are normal, but puberty does not occur (no estrogen).
• Female pseudohermaphroditism . Children with the 46.XX genotype (ovaries present) but usually have a male phenotype at birth. Increased sensitivity of the XX fetus to the effects of androgens during the critical period (8 weeks of intrauterine development) leads to the development of varying degrees of severity of labioscrotal fusion, the formation of the urogenital sinus and enlargement of the clitoris. Some babies appear male at birth with cryptorchidism •• Congenital adrenal hyperplasia. Defects leading to the development of female pseudohermaphroditism are deficiency of 21- and 11-hydroxylase, as well as 3--hydroxysterone dehydrogenase •• The influence of maternal androgens and progestin. As the potential danger to the developing fetus of drugs taken by the mother during pregnancy is realized, androgenic drugs are becoming an increasingly rare cause of female pseudohermaphroditism. Occasionally, the development of this pathology may be caused by a virilizing tumor or maternal illness during pregnancy. Careful collection of anamnestic data (pregnancy course, including drug intake and diseases) is necessary.
• Congenital defects of the external genitalia : hypospadias and microphallia •• Hypospadia of varying severity is recorded in isolation or in combination with other congenital defects, especially of the genitourinary system •• Microphallia. Boys' genitals are small, but well differentiated. There are standards for assessing the length of the erect penis, from early childhood to adulthood. Genital growth is determined by hormonal stimulation of the fetal testicles with pituitary gonadotropin ••• Etiology. Microphallia (penile hypoplasia) may indicate postnatal hypogonadotropic hypogonadism (as in Kallmann syndrome) or may reflect congenital hypopituitarism ••• Treatment. An effect can be expected from the use of testosterone (25–50 mg every 3 weeks for 3 months), which leads to a positive cosmetic effect without significantly accelerating skeletal maturation.
Management of a child with bisexual genitalia • A full diagnostic evaluation should be undertaken as soon as possible after the birth of a child with bisexual genitalia. It is necessary to convince parents to postpone giving the child a name and attributing gender until the end of the diagnostic measures. Needed: a thorough collection of family history, details of the course of pregnancy, general examination •• Examination. The size of the penis, the location of the urethra, the presence of palpable gonads (usually testes) and other signs (dysmorphic and asymmetric) should be assessed •• Laboratory studies include: chromosomal analysis, determination of electrolytes, testosterone, LH, FSH and 17-hydroxyprogesterone. Radiographic contrast examination of the urogenital sinuses often helps to detect the vagina and cervix, and sometimes it is possible to examine the fallopian tubes. Ultrasound of the pelvic organs can reveal the presence of the ovaries and uterus ••• With a karyotype of 46.XX and an increased content of 17-hydroxyprogesterone, 21-hydroxylase deficiency is most likely. With normal levels of 17-hydroxyprogesterone, true hermaphroditism is most likely. Assessment of the content of 11-deoxycortisol and dehydroepiandrosterone excludes the possibility of deficiency of 11-hydroxylase or 3-b-hydroxysterone dehydrogenase ••• With a karyotype of 46,XY, assessment of the content of sex and adrenal steroids before and after stimulation with ACTH and hCG allows us to identify rare forms of congenital adrenal hyperplasia and disorders synthesis and metabolism of testosterone ••• If glucocorticoid deficiency (including salt loss) is suspected, children should be closely monitored until test results are obtained ••• The diagnosis of partial androgen resistance depends on family history. High testosterone levels in newborns with elevated LH levels suggest androgen resistance.
• Gender assignment . To resolve many therapeutic and general issues, an accurate diagnosis is necessary •• Even noticeably virilized girls with 21 hydroxylase deficiency should be raised as girls, since with adequate treatment of the disease and cosmetic restoration of the external genitalia, they will have full reproductive capabilities •• For boys with a karyotype of 46, XY and bisexual genitalia, sex determination must be based on the decision of the ability to perform sexual functions as a man. This usually depends on the size of the penis and the surgeon's assessment of the possibility of surgical correction of hypospadias ••• Dysgenetic testicles and ovarian-testes should be removed, since the likelihood of their malignant transformation is high ••• To avoid unwanted hormonal influences during puberty, it is necessary to remove the gonads, not corresponding to gender.
• Treatment •• Adequate hormone replacement therapy, as a rule, should be prescribed during puberty •• Genetic counseling (including on issues of sexual behavior) of families and children is an important aspect of medical care •• Full, but tactful information about the results all tests (according to the patient's age) should lead to successful psychosexual adaptation •• Removal of the gonads. Due to possible malignancy, the XY gonad in resistant ovarian syndrome must be removed before puberty or immediately after diagnosis. There is no need to remove the gonads in patients with either Turner syndrome or Kallmann syndrome, since there is no potential for gonadal malignancy; with Turner syndrome hr. There is no Y, but in Kallmann syndrome the chromosomes are normal. Androgen insensitivity syndrome (see Testicular feminization syndrome) is characterized by the presence of type XY gonads; the gonads do not need to be removed before the completion of sexual development, since the risk of developing gonadal tumors is low until the age of 20 •• Sex change is carried out in case of hermaphroditism, as well as at the request of a transsexual, when the patient is convinced that his existing sexual characteristics do not correspond to his gender. After careful preoperative preparation (psychiatrist consultation, hormone replacement therapy), a destructive operation is performed, followed by organ reconstruction using various flaps and skin grafts.
Abbreviations. DHT - dihydrotestosterone, MIF - Mullerian inhibitory factor
ICD-10. Q56 Sex ambiguity and pseudohermaphroditism
Note. Hermaphroditism (from Greek mythology - Hermaphroditos, the son of the god Hermes [Hermes] and the goddess Aphrodite [Aphrodite]; while swimming, he merged his body with a nymph) - the presence of both male and female reproductive glands in an individual (true hermaphroditism) "androgyny" bisexuality • • The term is also (and from a genetic point of view - loosely) used in the meaning: the presence in an individual of characteristics of both sexes (including behavioral) “ambisexuality” bisexuality “bisexuality” intersexism “intersexuality.