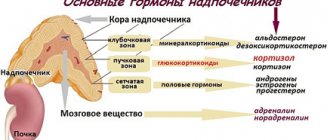
Адреногенитальный синдром (АГС) – это комплекс нарушений в процессе выработки кортикостероидов.
Подобные нарушения передаются по наследству, по аутосомно-рецессивному типу, поэтому синдром может встречаться у представителей обоих полов с одинаковой частотой. Вероятность рождения ребенка с таким синдромом при носительстве патологического гена у обоих родителей достигает 25%, в браке носителя и больного – 75%. Если один из родителей имеет полноценные ДНК, клинические проявления синдрома у детей не развиваются. При наличии АДС у отца и матери ребенок также будет болен. Примерно 1 из 10000 детей рождается с данной патологией.
Адреногенитальный синдром и его формы
В 90% случаев развитие болезни связано с отсутствием фермента гидроксилазы. Это ведет к тяжелым нарушениям синтеза гормонов. Врожденный адреногенитальный синдром встречается у 1 из 5000 новорожденных. Заболевание сопровождается усилением активности коры надпочечников и увеличением концентрации в крови мужских половых гормонов – андрогенов.
Кроме ферментативных нарушений, в развитии патологии может иметь значение опухоль надпочечников. Она ведет к развитию приобретенного варианта заболевания.
Формы адреногенитального синдрома:
- Сольтеряющая характеризуется полным отсутствием у детей 21-гидроксилазы и выраженными нарушениями солевого обмена. Уже в первые дни жизни у новорожденного наблюдается обильная рвота, недостаточность кровообращения, потеря веса тела. Последующее обезвоживание приводит к серьезным нарушениям обмена калия и натрия, кислотно-щелочного равновесия.
- Вирильная форма сопровождается развитием мужских половых признаков у девочек. Несмотря на «женский» генетический набор наружные половые органы родившихся девочек в той или иной мере напоминают мужские. Это так называемый ложный женский гермафродитизм.
- Вирильная форма у мальчиков сразу после рождения не распознается, их гениталии имеют соответствующий полу вид. К 5 – 7 годам у них появляются признаки полового созревания, на основании чего и предполагают этот диагноз.
- Пубертатная форма проявляется у подростков. У девочек отмечается недоразвитие молочных желез, увеличение клитора, позднее наступление первой менструации, удлинение менструального цикла, гирсутизм, угревая сыпь. У мальчиков идет процесс преждевременного физического и полового развития.
- Постпубертатная форма развивается у женщин, нередко после прервавшейся беременности. Менструальный цикл удлиняется, месячные становятся скудными. Гирсутизм слабо выражен.
В классификации также представлены более редкие гипертензивная форма с задержкой в организме воды и солей и развитием стойкой гипертензии, липидная и гипертермическая формы.
Адреногенитальный синдром, или врожденная дисфункция коры надпочечников, — это группа врожденных моногенных аутосомно-рецессивных заболеваний. Первое описание пациента с данным синдромом относится к середине XIX века. В 1865 г. итальянский анатом Luigi De Crecchio описал пациента мужского пола, который умер при явлениях «адинамии и рвоты», при вскрытии у него были обнаружены внутренние женские половые органы (L. de Crecchio: Sopra un caso di apparenze virili in una donna. Napoli, Morgagni, 1865, 7: 151.) [1]. Уже в конце XIX века была выявлена взаимосвязь женского псевдогермафродитизма с гиперплазией надпочечников. В начале XX века появились сообщения о связи преждевременного полового развития у мальчиков с гиперплазией, или объемными образованиями надпочечников. Сочетание в одной семье девочек с гермафродитизмом и мальчиков с преждевременным половым развитием позволило ряду авторов предположить единую врожденную причину данных состояний (J. Fibiger: Beiträge zur Kenntniss des weiblichen Scheinzwittertums. Virchows Archiv für pathologische Anatomie und Physiologie, und für klinische Medizin, Berlin, 1905, 181: 1-51. R. Debré, G. Sémélaigne: Hypertrophie considerable des capsules surrénales chez un nourrison mort a 10 mois sans avoir augmenté de poids depuis sa naissance. Bull Soc Pediat, Paris, 1925, 23: 270—271) [2]. В 1912 г. A. Gallais впервые ввел термин адреногенитальный синдром [3].
В отечественной литературе О.В. Верещинский в 1924 г. впервые обобщил 12 случаев надпочечно-полового синдрома [4]. В работах 1920—1930-х гг. одного из основателей советской педиатрии В.И. Молчанова и выдающегося патолога И.В. Давыдовского имеются попытки объяснить причину адреногенитального синдрома [5]. В 1946 г. один из основателей эндокринологии в России директор Всесоюзного института экспериментальной эндокринологии (в последующем ЭНЦ) Н.А. Шерешевский выделил мышечный тип супрарено-генитального синдрома, который в современной классификации соответствует вирильной форме дефицита 21-гидроксилазы [6].
Единственным способом лечения как девочек с явлениями вирилизма, так и мальчиков с преждевременным половым развитием при адреногенитальном синдроме был хирургический. Удаление одного надпочечника давало частичный и временный эффект, в то время как двусторонняя адреналэктомия была невозможна из-за отсутствия на тот момент заместительной терапии [6].
Развитие биохимии и фармакологии положило начало новому этапу в изучении адреногенитального синдрома. Синтез и клиническое применение кортикостероидных препаратов, за которое P. Hench и E. Kendall в 1949 г. получили Нобелевскую премию, позволило эндокринологам не только установить этиологию и патогенез адреногенитального синдрома, но и проводить патогенетическую терапию данного заболевания. F. Bartter и L. Wilkins первыми связали адреногенитальный синдром с дефицитом ферментов, обеспечивающих синтез кортизола [7, 8]. Начиная c 1950-х годов, сформулировано современное определение врожденной дисфункции коры надпочечников (ВДКН) как группы заболеваний с аутосомно-рецессивным типом наследования, в основе которых лежит дефект одного из ферментов или транспортных белков, принимающих участие в биосинтезе кортизола в коре надпочечников. Снижение биосинтеза кортизола, согласно классическому принципу обратной связи, приводит к повышению секреции АКТГ и, как следствие, развитию гиперплазии коры надпочечников и накоплению метаболитов, предшествующих дефектному этапу стероидогенеза. При дефиците 21-гидроксилазы не нарушен синтез надпочечниковых андрогенов, что и обусловливает клиническую картину заболевания: внутриутробную вирилизацию плодов женского пола, проявляющуюся гермафродитным строением наружных гениталий. После рождения избыток андрогенов приводит к дальнейшей вирилизации у девочек и ложному преждевременному изосексуальному половому развитию у мальчиков. Наблюдается ускорение роста, преждевременное половое оволосение. Быстрое закрытие зон роста приводит к выраженной конечной низкорослости. Дефицит минералокортикоидов, встречающийся в 75% случаев, проявляется синдромом потери соли и без лечения приводит к гибели пациентов в неонатальный период.
С 1951 г. в лечении ВДКН с хорошим эффектом применяют кортикостероидные препараты. Глюкокортикоиды позволяют не только заместить дефицит кортизола, имеющийся при данном заболевании, но и снизить уровень АКТГ, тем самым нормализуется повышенный уровень андрогенов, вызванный избыточной стимуляцией надпочечников [8, 9]. Синтез стероидов с минералокортикоидной активностью позволил проводить также заместительную терапию при дефиците альдостерона, что обеспечило выживаемость пациентов с сольтеряющей формой заболевания.
Развитие биохимии в 1950—1960-х гг. обеспечило новую ступень в эволюции наших представлений об адреногенитальном синдроме. Использование метода хроматографии, предложенного еще в начале XX века, успешно развивалось в 1950-х г. и нашло широкое применение для разделения стероидных соединений, выделенных из крови пациентов с ВДКН. В Институте экспериментальной эндокринологии и химии гормонов АМН СССР (в последующем ЭНЦ) под руководством директора акад. Н.А. Юдаева было создано целое подразделение, занимающееся биохимией стероидных соединений, были разработаны методы определения стероидных гормонов и их метаболитов [10]. На основании лабораторных исследований совместно с врачами-клиницистами были определены особенности секреции стероидных гормонов при различных эндокринных заболеваниях.
Количественное определение в крови пациентов различных стероидных соединений в совокупности с клиническими данными позволило описать новые варианты ферментативных дефектов, приводящих к ВДКН. Так, в 1956 г. W. Eberlein и A. Bongiovanni [11] связали описанную ранее гипертоническую форму адреногенитального синдрома с дефицитом 11b-гидроксилазы. При данной форме ВДКН отмечается избыток андрогенов, клинически проявляющийся так же, как и при дефиците 21-гидроксилазы, и избыток 11-дезоксикортикостерона, обладающего минералокортикоидной активностью и вызывающий низкорениновую артериальную гипертензию.
В 1957 г. A. Prader и R. Siebenmann [12] описали семью с липоидной гиперплазией надпочечников, самой тяжелой формой ВДКН, при которой тотальный дефицит всех стероидных гормонов приводит к развитию сольтеряющего синдрома, ложного мужского гермафродитизма и первичного гипогонадизма как у мужчин, так и у женщин. В 1960 г. A. Bongiovanni описал пациентов с дефицитом 3b-гидроксистероиддегидрогеназы. При данной форме ВДКН помимо сольтеряющего синдрома имеет место нарушение строения наружных гениталий как при кариотипе 46ХХ, так и при 46XY. Это связано с повышенным уровнем дигидроэпиандростерона, являющегося слабым андрогеном [13].
И, наконец, в 1966 г. E. Biglieri [14] описал пациента с последней (по числу ферментов, участвующих в синтезе кортизола) формой ВДКН — дефицитом 17a-гидроксилазы, при которой имеется дефицит андрогенов и избыток минералокортикоидов. У пациентов отмечается ложный мужской гермафродитизм, первичный гипогонадизм и низкорениновая артериальная гипертензия с гипокалиемией.
В СССР на базе Института экспериментальной эндокринологии и химии гормонов АМН СССР (в последующем ЭНЦ) в 1961 г. была организована детская эндокринологическая клиника под руководством проф. М.А. Жуковского. В детской клинике активно проводилась клиническая и научная работа по адреногенитальному синдрому [2, 15, 17, 18]. В 1969 г. состоялся III Всесоюзный съезд детских врачей, на котором были озвучены актуальные вопросы детской эндокринологии [16, 19]. По результатам этого съезда, детская эндокринология была выделена в отдельную специальность. Уже в июне 1970 г. в Иваново была проведена I Всесоюзная конференция педиатров-эндокринологов, на которой обсуждались наиболее актуальные проблемы диагностики и лечения эндокринных заболеваний у детей, несколько докладов были посвящены врожденной дисфункции коры надпочечников [20].
В 1977 г. впервые стал доступен простой и надежный метод скрининга 21-гидроксилазной недостаточности, основанный на исследовании 17-гидроксипрогестерона радиоиммунным методом [21]. Первый пилотный проект неонатального скрининга был осуществлен на Аляске в популяции эскимосов, отличающейся высокой частотой данной патологии. По результатам данного проекта, частота 21-гидроксилазной недостаточности среди эскимосов Аляски составила 1:280 новорожденных и в настоящее время остается самой высокой частотой данной патологии в мире [22]. Внедрение неонатального скрининга позволило устанавливать диагноз и начинать лечение пациента до начала развития сольтеряющего криза, что значительно снизило летальность при данном заболевании. Кроме того, с помощью скрининга стала возможной диагностика вирильной формы заболевания у мальчиков в неонатальный период, что существенно улучшает ростовой прогноз этих пациентов. И, наконец, у девочек с выраженной вирилизацией гениталий при рождении благодаря процедуре скрининга не возникает ошибок с выбором половой принадлежности. К 1991 г. неонатальный скрининг на адреногенитальный синдром проводился в 13 странах мира [23]. В России процедура скрининга внедрена с 2006 г. Внедрение проекта осуществлялось ФГУ Эндокринологическим научным центром (директор — акад. И.И. Дедов), как головным эндокринологическим учреждением Министерства здравоохранения и социального развития РФ.
Широкое внедрение неонатального скрининга открыло новые возможности изучения эпидемиологии ВДКН. К середине XX века в США впервые была оценена распространенность адреногенитального синдрома как 1 случай на 67 000 новорожденных [24]. При этом подавляющее большинство пациентов имело вирильную или неклассическую форму дефицита 21-гидроксилазы, поскольку без лечения пациенты с сольтеряющей формой умирали до постановки диагноза. В настоящее время частота встречаемости только классических форм дефицита 21-гидроксилазы, рассчитанная на основании данных неонатального скрининга в мировой популяции, составляет 1 случай на 14 199 новорожденных [23]. При этом около 75% случаев приходится на сольтеряющую форму и 25% на простую вирильную. Распространенность же неклассической формы дефицита 21-гидроксилазы еще выше, в европейской популяции достигает 1 случая на 1000.
В 1980-е гг. началась новая эра в изучении адреногенитального синдрома, связанная с успехами молекулярной генетики. Были клонированы гены, кодирующие ферменты стероидогенеза, и разработана методика молекулярной диагностики для всех форм ВДКН [25—28]. Неожиданностью оказалось отсутствие мутаций в гене CYP11A1 у пациентов с липоидной гиперплазией надпочечников. Дальнейшие исследования доказали, что в большинстве случаев к липоидной гиперплазии надпочечников приводит дефект STAR-протеина, обеспечивающего транспорт холестерина на внутреннюю мембрану митохондрий, где он становится доступен для воздействия фермента 11a-гидроксилазы [29].
Разработка молекулярно-генетических методов обследования пациентов позволила проводить пренатальную диагностику адреногенитального синдрома. Вскоре французскими и американскими учеными была предложена пренатальная терапия дексаметазоном, целью которой было предотвратить внутриутробную вирилизацию плодов женского пола при дефиците 21-гидроксилазы и 11b-гидроксилазы [30, 31]. Дексаметазон не подвергается воздействию фермента 11b-гидроксистероиддегидрогеназы 2-го типа в плаценте, следовательно, поступает к плоду, по механизму отрицательной обратной связи снижает уровень АКТГ, препятствуя тем самым избыточной секреции надпочечниковых андрогенов, и предотвращает вирилизацию наружных гениталий у плода женского пола.
В 2004 г. группой Британских ученых открыта еще одна новая форма ВДКН — дефицит оксидоредуктазы, вызванная дефектом гена POR [32]. Пациенты с клинической картиной сочетанного дефицита P450C17 и P450C21 были описаны еще в 1985 г., но в генах этих ферментов у них не обнаруживались мутации, и механизм развития таких проявлений никто не мог объяснить до 2004 г. Цитохром-Р450-оксидоредуктаза обеспечивает электронами все микросомальные ферменты Р450, включая CYP51A1, CYP17A1, CYP21A2, и CYP19A1, поэтому клиническая картина отражает сочетанный дефицит этих ферментов. Относительная недостаточность 21-гидроксилазы (CYP21A2) приводит к внутриутробной вирилизации плодов с кариотипом 46ХХ; дефицит 17a-гидроксилазы (CYP17A1) может приводить к недостаточной маскулинизации плодов с кариотипом 46XY и к развитию андрогеновой недостаточности в пубертатный период; недостаточность CYP51A1 может приводить к формированию костных аномалий, таких как краниосиностоз, синостоз лучевой и локтевой костей и множественные контрактуры; недостаточность ароматазы (CYP19A1) может вызывать гипогонадизм у женщин, а недостаточность плацентарной ароматазы приводит к вирилизации матери во время беременности. Описание дефицита оксидоредуктазы доказывает, что рано ставить точку в описании механизмов развития нарушений стероидогенеза в надпочечниках.
В последнее десятилетие основные усилия врачей были направлены на оптимизацию терапии адреногенитального синдрома, длительное катамнестическое наблюдение выявило осложнения и недостатки стандартных схем терапии. В дополнение к глюкокортикоидной терапии было предложено использование препаратов гормона роста в сочетании с пролонгированными аналогами люлиберина с целью улучшения ростового прогноза пациентов [33]. В стадии клинической апробации находятся новые глюкокортикоидные препараты с измененными фармакодинамическими характеристиками [34].
Приведенные данные описывают эволюцию наших представлений о механизмах возникновения ВДКН и методах лечения пациентов с этой патологией надпочечников. В прошлом редкое, трудно распознаваемое заболевание стало объектом популяционного скрининга во многих странах, в том числе и в России. Конечно, в ближайшем будущем предстоит решить много организационных задач, связанных с проведением скрининга, обеспечением квалифицированной помощью пациентов, проживающих в отдаленных от крупных центров регионах страны. Наше ближайшее «завтра» с помощью современной организации здравоохранения и высокой квалификации эндокринологов должно избавить общество от ошибочного или пропущенного диагноза ВДКН у новорожденных, обеспечить этим пациентам полноценное физическое и психологическое развитие и возможность репродукции.
Но помимо этих административных и экономических задач, несомненно имеющих государственное значение для России, остается много вопросов, которые предстоит решать ученым и врачам всего мира. Следует отметить, что по-прежнему определенная категория пациентов не соответствует в полной мере критериям диагностики ныне известных вариантов дефектов стероидогенеза в надпочечниках. Подтверждением тому является открытие дефекта оксидоредуктазы. И эта новая форма, которая недавно была выделена в группе ВДКН, вероятно, не является последней. Врачи-клиницисты, описывая и публикуя необычные наблюдения, постепенно формируют гипотезы о новых, еще не изученных ферментах и коферментах стероидогенеза, которые могут приводить к аналогичным нарушениям. Вопросы о схемах лечения, адаптации пациентов к мужскому или женскому полу в разных возрастных группах, вопросы репродукции, дородовой диагностики и лечения — все эти темы по-прежнему являются предметом жарких споров, а новые исследования приводят новые аргументы.
Если говорить о будущем, может быть уже о «послезавтрашнем» дне, нельзя обойти стороной тему, которая будоражит умы ученых и врачей уже не первое десятилетие — «генная терапия». Генетика — наука настоящего и будущего, на которую возлагают надежды в лечении многих заболеваний. Первая публикация, посвященная генотерапии наследственных болезней, вышла в 1972 г. в журнале Science («Gene therapy for human genetic disease?») [35]. В 1990 г. в Национальном институте здоровья США этот метод был впервые применен для лечения врожденного иммунодефицита у людей (наблюдался временный эффект). Но с тех пор метод так и не стал общедоступным и широко применяемым, поскольку его применение связано с множеством технических и этических сложностей. Дефекты генов ферментов стероидогенеза пока не стали объектом генотерапии, однако уже очень широко внедрен метод дородовой и предимплантационной диагностики ВДКН, который позволяет существенно повысить вероятность рождения здорового ребенка с помощью методов экстракорпорального оплодотворения [36].
Заключение
ВДКН — одно из многих наследственных заболеваний, которое уже не относится к разряду «редких». Всеми признано огромное социальное значение этой проблемы в здравоохранении. Значительные успехи мировой науки дают нам возможность сделать людей с этой патологией нормальными членами общества, способными к обучению, труду и деторождению. В России диагностика и лечения ВДКН осуществляется на современном уровне, имеется большой опыт в крупнейших эндокринологических центрах страны, внедрен скрининг новорожденных. Ближайшая первостепенная задача эндокринологического сообщества России — обеспечить современной квалифицированной помощью всех пациентов. Велика роль и научных исследований в этой области в России как клинических, так и экспериментальных, которые внесут свой вклад в мировую эндокринологию.
Причины
Причины адреногенитального синдрома связаны с генетическим нарушением – дефектом одного из генов 6-1 пары хромосом. Этот ген рецессивный, и проявляется только в случае рождения ребенка с обеими измененными хромосомами. Его родители обязательно должны быть либо носителями патологического гена, либо страдать этим заболеванием.
Этот ген отвечает за синтез фермента 21-гидроксилазы. Ее недостаток или отсутствие приводят к компенсаторному усиленному синтезу гормона гипофиза АКТГ, что ведет к гипертрофии надпочечников и к усилению выработки андрогенов. Они подавляют функцию яичников и образование женских половых гормонов – эстрогенов.
Почему происходит мутация этого гена, неизвестно. Носителями измененного гена с равной частотой могут быть и мальчики, и девочки. Клиническое развитие заболевания тоже не зависит ни от пола ребенка, ни от того, кто из родителей болен, а кто лишь передал патологический ген.
2.Причины
Как указано выше, непосредственной причиной развития адреногенитального синдрома является обусловленный генетической мутацией дефицит белка 21-гидроксилазы. Различают три основные формы:
- с сольтеряющим синдромом (полное отсутствие фермента);
- простая вирильная форма (частичный дефицит 21-гидроксилазы);
- неклассическая постпубертатная форма.
Первый вариант встречается наиболее часто; его доля в общем объеме случаев адреногенитального синдрома достигает 75%.
Приобретенная постпубертатная форма, развивающаяся уже в зрелом возрасте, чаще всего обусловлена онкопроцессом в коре надпочечников.
Посетите нашу страницу Эндокринология
Симптомы и диагностика заболевания
Больные должны наблюдаться у детского эндокринолога. Грамотное лечение именно у этого специалиста дает возможность нормализовать гормональную функцию.
Симптомы адреногенитального синдрома:
- быстрый рост в подростковом возрасте;
- возникновение у девушек явлений гирсутизма – волос на верхней губе, внутренней поверхности бедер, по белой линии живота, вокруг сосков;
- выраженная угревая сыпь;
- нарушения менструального цикла, бесплодие.
Внешние признаки адреногенитального синдрома наиболее заметны у девушек:
- мужской тип телосложения – высокий рост, узкий таз, недоразвитые молочные железы, широкие плечи; вес в пределах нормы;
- нарушение строения наружных половых органов при нормально сформированной матке и естественной для женщины сексуальной ориентации;
- при наступлении беременности частое осложнение – прерывание в 1-м триместре.
Диагностика адреногенитального синдрома проводится с помощью таких исследований:
- определение уровня гормонов: тестостерона, ДЭА, ДЭА-С, 17-ОНП, ЛГ, ФСГ, показывающих степень нарушения функций надпочечников и яичников;
- пробы с дексаметазоном и АКТГ, необходимые для дифференциальной диагностики с синдромом поликистозных яичников;
- УЗИ яичников;
- определение длительности фаз цикла путем составления графика базальной температуры.
Диагностику адреногенитального синдрома необходимо доверить квалифицированному специалисту. Обязательное условие – наличие современной лаборатории для гормональных исследований. Все эти услуги предлагает своим пациентам клиника «Мама Папа Я».
Что будет, если не лечить АГС
Переизбыток андрогенов ведет к неправильному развитию половых органов у девочек. Нарушения варьируются – от легкого увеличения клитора до выраженных признаков, свойственных мужчинам. В тяжелых случаях пол ребенка сложно установить, поскольку клитор похож на пенис, а отверстие уретры находится на его головке.
Неправильное развитие больших половых губ придает им сходство с мошонкой. Внутренние половые органы при этом заболевании развиты по женскому типу, но иногда пол ребенка можно определить только на УЗИ.
В дальнейшем у девочек появляются другие мужские признаки:
- низкий голос;
- мускулистое телосложение;
- оволосение по мужскому типу, намного опережающее возрастное. К четырем годам волосы начинают появляться под мышками, а в 8-9 лет – на лице.
К началу подросткового периода девочки имеют специфическое телосложение:
- низкорослость, вызванная быстрым закрытием зон костного роста;
- крупное туловище с короткими конечностями;
- плечевая область шире тазовой;
- неразвитые молочные железы.
При легких формах адреногенитального синдрома менструации есть, но они начинаются позже, болезненны (альгоменорея) и довольно скудны. При тяжелых формах менструации полностью отсутствуют.
Если не лечить АГС, у женщины на всю жизнь останутся серьезные проблемы: она будет иметь мужские черты, но внутреннее женское строение. При этом она никогда не станет матерью.
Лечение
При сольтеряющей форме заболевания лечение начинают сразу после рождения ребенка, как только появились первые симптомы. Оно включает интенсивную терапию с введением жидкости, солей, гормональных препаратов.
Лечение адреногенитального синдрома включает:
- длительный прием гормонов надпочечников – глюкокортикоидов, который должен начинаться в раннем детстве (до 7 лет);
- по показаниям – назначение женских половых гормонов, особенно при подготовке к беременности;
- при необходимости еще в детском возрасте пациенткам проводят хирургическую пластику наружных половых органов.
Значительную трудность вызывает сохранение беременности у больных женщин. Однако грамотный гинеколог-эндокринолог способен подобрать необходимые лекарственные препараты, чтобы пациентка смогла родить здорового ребенка.
Симптомы дисфункции коры надпочечников
При врожденном варианте адреногенитального синдрома фигура пациенток уже в детском возрасте приобретает мужские черты. У них рано закрываются зоны роста в костях, поэтому они имеют невысокий рост, грубеет голос, половые органы имеют неправильное строение (гипертрофированный клитор, видоизмененные половые губы напоминают мошонку).
Кроме классической формы АГС существуют :
- пубертатная, когда симптомы проявляются в переходном подростковом возрасте, в период физиологического усиления гормональной функции коры надпочечников;
- постпубертатная, когда проявления манифестируют в конце второго десятилетия жизни, часто после самопроизвольного выкидыша на раннем сроке беременности, неразвивающейся беременности или после медицинского аборта;
- латентная форма, единственным симптомом которой может быть невынашивание беременности на ранних сроках.
При исследовании кариотипа определяется 46 ХХ хромосом (женский набор). Внутренние половые органы также соответствуют женскому организму (матка, яичники). Половое созревание наступает рано (в 6-7 лет): оволосение развивается по мужскому типу, молочные железы слабо развиты, нередко отсутствуют менструации, однако, может присутствовать полноценный менструальный цикл. В дальнейшем пациенток с таким диагнозом ожидает бесплодие или привычное невынашивание плода.
Если же нарушение функции коры надпочечников вторично, пациентки жалуются на:
- внезапно появившееся избыточное оволосение (гирсутизм);
- нерегулярность менструального цикла;
- невозможность забеременеть;
- уменьшение молочных желез;
- увеличение размера клитора.
При выполнении УЗИ внутренних органов диагностируют недоразвитость или атрофию матки и яичников.
В обоих случаях патология может развиваться с преобладанием одного из синдромов:
- Сольтеряющий: пациентки отмечают общую вирилизацию (омужествление), гипотонию (сниженное артериальное давление), отеки.
- Гипертензивный: проявляется поражением миокарда и сосудов, повышением артериального давления.
- Вирилизация: фигура и голос пациенток становятся более мужскими, однако поражение внутренних органов отсутствует.
Записаться на прием
Почему необходимо обратиться в клинику «Мама Папа Я»
Сеть семейных клиник «Мама Папа Я» расположена в Москве и других городах. При адреногенитальном синдроме оказываются такие медицинские услуги по доступной цене:
- лабораторная диагностика гормональных нарушений на современном оборудовании;
- прием профильных специалистов – педиатра, гинеколога, эндокринолога;
- последовательное наблюдение за ребенком, преемственность в лечении у докторов разного профиля;
- современные методы лечения при вынашивании беременности;
- назначение лекарственной терапии по современным методикам.
Врачи нашей клиники проводят раннюю диагностику и назначают грамотное лечение при любой форме заболевания. В таких условиях проявления адреногенитального синдрома хорошо корректируются. Записывайтесь на прием прямо сейчас!
Отзывы
Хорошая клиника, хороший врач! Раиса Васильевна может понятно и доступно объяснить, в чем суть проблемы. Если что-то не так, она обо всем говорить прямо, не завуалированно, как это порой делают другие врачи. Не жалею, что попала именно к ней.
Анна
Хочу выразить благодарность работникам клиники Мама, Папа, я. В клинике очень дружелюбная атмосфера, очень приветливый и веселый коллектив и высококвалифицированные специалисты. Спасибо вам большое! Желаю процветания вашей клинике.
Анонимный пользователь
Сегодня удаляла родинку на лице у дерматолога Кодаревой И.А. Доктор очень аккуратная! Корректная! Спасибо большое! Администратор Борщевская Юлия доброжелательна, четко выполняет свои обязанности.
Белова Е.М.
Сегодня обслуживалась в клинике, осталась довольна персоналом, а также врачом гинекологом. Все относятся к пациентам с уважением и вниманием. Спасибо им большое и дальнейшего процветания.
Анонимный пользователь
Клиника «Мама Папа Я» в Люберцах очень хорошая. Коллектив дружелюбный, отзывчивый. Советую данную клинику всем своим знакомым. Спасибо всем врачам и администраторам. Клинике желаю процветания и много адекватных клиентов.
Иратьев В.В.
Посетили с ребенком Клинику «Мама Папа Я». Нужна была консультация детского кардиолога. Клиника понравилась. Хороший сервис, врачи. В очереди не стояли, по стоимости все совпало.
Евгения
Первое посещение понравилось. Меня внимательно осмотрели, назначили дополнительные обследования, дали хорошие рекомендации. Буду продолжать лечение и дальше, условия в клинике мне понравились.
Кристина
Доктор внимательно осмотрела моего мужа, назначила ЭКГ и поставили предварительный диагноз. Дала рекомендации по нашей ситуации и назначила дополнительное обследование. Пока замечаний нет. Финансовые договоренности соблюдены.
Марина Петровна
Очень понравилась клиника. Обходительный персонал. Была на приеме у гинеколога Михайловой Е.А. осталась довольна, побольше таких врачей. Спасибо!!!
Ольга