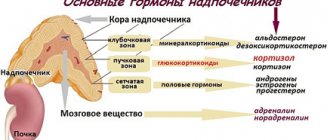
Adrenogenital syndrome (AGS) is a complex of disorders in the production of corticosteroids.
Such disorders are inherited in an autosomal recessive manner, so the syndrome can occur in both sexes with equal frequency. The probability of having a child with this syndrome when both parents are carriers of the pathological gene reaches 25%, in a marriage between a carrier and a patient – 75%. If one of the parents has complete DNA, clinical manifestations of the syndrome do not develop in children. If the father and mother have ADS, the child will also be sick. Approximately 1 in 10,000 children are born with this pathology.
Adrenogenital syndrome and its forms
In 90% of cases, the development of the disease is associated with the absence of the hydroxylase enzyme. This leads to severe disruption of hormone synthesis. Congenital adrenogenital syndrome occurs in 1 in 5,000 newborns. The disease is accompanied by increased activity of the adrenal cortex and an increase in the concentration of male sex hormones - androgens - in the blood.
In addition to enzymatic disorders, adrenal tumors may play a role in the development of pathology. It leads to the development of an acquired variant of the disease.
Forms of adrenogenital syndrome:
- Salt-wasting disease is characterized by a complete absence of 21-hydroxylase in children and severe disorders of salt metabolism. Already in the first days of life, the newborn experiences profuse vomiting, circulatory failure, and loss of body weight. Subsequent dehydration leads to serious disturbances in potassium and sodium metabolism and acid-base balance.
- The virile form is accompanied by the development of male sexual characteristics in girls. Despite the “female” genetic makeup, the external genitalia of born girls are more or less reminiscent of male ones. This is the so-called false female hermaphroditism.
- The virile form in boys is not recognized immediately after birth; their genitals have a gender-appropriate appearance. By the age of 5–7 years, they show signs of puberty, on the basis of which this diagnosis is assumed.
- The pubertal form manifests itself in adolescents. In girls, there is underdevelopment of the mammary glands, enlargement of the clitoris, late onset of the first menstruation, prolongation of the menstrual cycle, hirsutism, and acne. Boys are undergoing a process of premature physical and sexual development.
- The postpubertal form develops in women, often after an interrupted pregnancy. The menstrual cycle lengthens, periods become scanty. Hirsutism is mild.
The classification also presents the rarer hypertensive form with retention of water and salts in the body and the development of persistent hypertension, lipid and hyperthermic forms.
Adrenogenital syndrome, or congenital dysfunction of the adrenal cortex, is a group of congenital monogenic autosomal recessive diseases. The first description of a patient with this syndrome dates back to the mid-19th century. In 1865, the Italian anatomist Luigi De Crecchio described a male patient who died with symptoms of “adynamia and vomiting”; an autopsy revealed internal female genitalia (L. de Crecchio: Sopra un caso di apparenze virili in una donna. Napoli, Morgagni, 1865, 7: 151.) [1]. Already at the end of the 19th century, the relationship between female pseudohermaphroditism and adrenal hyperplasia was identified. At the beginning of the 20th century, reports appeared about the connection between premature puberty in boys and hyperplasia, or space-occupying formations of the adrenal glands. The combination of girls with hermaphroditism and boys with premature sexual development in one family allowed a number of authors to suggest a single congenital cause of these conditions (J. Fibiger: Beiträge zur Kenntniss des weiblichen Scheinzwittertums. Virchows Archiv für pathologische Anatomie und Physiologie, und für klinische Medizin, Berlin, 1905 , 181: 1-51. R. Debré, G. Sémélaigne: Hypertrophie significant des capsules surrénales chez un nourrison mort a 10 mois sans avoir augmenté de poids depuis sa naissance. Bull Soc Pediat, Paris, 1925, 23: 270-271) [2]. In 1912, A. Gallais first introduced the term adrenogenital syndrome [3].
In domestic literature O.V. Vereshchinsky in 1924 first summarized 12 cases of adrenal-genital syndrome [4]. In works of the 1920-1930s. one of the founders of Soviet pediatrics V.I. Molchanov and the outstanding pathologist I.V. Davydovsky there are attempts to explain the cause of adrenogenital syndrome [5]. In 1946, one of the founders of endocrinology in Russia, director of the All-Union Institute of Experimental Endocrinology (later ENC) N.A. Shereshevsky identified a muscular type of suprarenogenital syndrome, which in the modern classification corresponds to the viril form of 21-hydroxylase deficiency [6].
The only treatment for both girls with virilism and boys with precocious puberty due to adrenogenital syndrome was surgery. Removal of one adrenal gland gave a partial and temporary effect, while bilateral adrenalectomy was impossible due to the lack of replacement therapy at that time [6].
The development of biochemistry and pharmacology marked the beginning of a new stage in the study of adrenogenital syndrome. The synthesis and clinical use of corticosteroid drugs, for which P. Hench and E. Kendall received the Nobel Prize in 1949, allowed endocrinologists not only to establish the etiology and pathogenesis of adrenogenital syndrome, but also to carry out pathogenetic therapy for this disease. F. Bartter and L. Wilkins were the first to associate adrenogenital syndrome with a deficiency of enzymes that provide cortisol synthesis [7, 8]. Since the 1950s, the modern definition of congenital adrenal cortex dysfunction (CAD) has been formulated as a group of diseases with an autosomal recessive pattern of inheritance, which are based on a defect in one of the enzymes or transport proteins involved in the biosynthesis of cortisol in the adrenal cortex. A decrease in cortisol biosynthesis, according to the classical feedback principle, leads to increased ACTH secretion and, as a consequence, the development of adrenal hyperplasia and the accumulation of metabolites that precede the defective stage of steroidogenesis. With 21-hydroxylase deficiency, the synthesis of adrenal androgens is not impaired, which determines the clinical picture of the disease: intrauterine virilization of female fetuses, manifested by the hermaphroditic structure of the external genitalia. After birth, excess androgens lead to further virilization in girls and false premature isosexual sexual development in boys. Acceleration of growth and premature pubertal hair growth are observed. Rapid closure of growth zones leads to pronounced final short stature. Mineralocorticoid deficiency, which occurs in 75% of cases, manifests itself as salt wasting syndrome and, without treatment, leads to the death of patients in the neonatal period.
Since 1951, corticosteroid drugs have been used in the treatment of CAH with good effect. Glucocorticoids can not only replace the cortisol deficiency present in this disease, but also reduce the level of ACTH, thereby normalizing the increased level of androgens caused by excessive stimulation of the adrenal glands [8, 9]. The synthesis of steroids with mineralocorticoid activity also made it possible to carry out replacement therapy for aldosterone deficiency, which ensured the survival of patients with the salt-wasting form of the disease.
Development of biochemistry in the 1950-1960s. provided a new step in the evolution of our ideas about adrenogenital syndrome. The use of the chromatography method, proposed at the beginning of the 20th century, was successfully developed in the 1950s and found widespread use for the separation of steroid compounds isolated from the blood of patients with CAI. At the Institute of Experimental Endocrinology and Hormone Chemistry of the USSR Academy of Medical Sciences (later ENC), under the leadership of the director, academician. ON THE. Yudaev, an entire department was created dedicated to the biochemistry of steroid compounds; methods for determining steroid hormones and their metabolites were developed [10]. Based on laboratory studies, together with clinicians, the characteristics of the secretion of steroid hormones in various endocrine diseases were determined.
Quantitative determination of various steroid compounds in the blood of patients in combination with clinical data made it possible to describe new variants of enzymatic defects leading to CAI. Thus, in 1956, W. Eberlein and A. Bongiovanni [11] associated the previously described hypertensive form of adrenogenital syndrome with 11b-hydroxylase deficiency. With this form of CDCN, there is an excess of androgens, clinically manifested in the same way as with 21-hydroxylase deficiency, and an excess of 11-deoxycorticosterone, which has mineralocorticoid activity and causes low-renin arterial hypertension.
In 1957, A. Prader and R. Siebenmann [12] described a family with lipoid adrenal hyperplasia, the most severe form of CAH, in which a total deficiency of all steroid hormones leads to the development of salt-wasting syndrome, false male hermaphroditism and primary hypogonadism in both men and women. and in women. In 1960, A. Bongiovanni described patients with 3b-hydroxysteroid dehydrogenase deficiency. With this form of CAI, in addition to salt-wasting syndrome, there is a disturbance in the structure of the external genitalia with both karyotypes 46XX and 46XY. This is due to increased levels of dihydroepiandrosterone, which is a weak androgen [13].
And finally, in 1966, E. Biglieri [14] described a patient with the last (in terms of the number of enzymes involved in the synthesis of cortisol) form of CAH—17a-hydroxylase deficiency, in which there is a deficiency of androgens and an excess of mineralocorticoids. Patients have false male hermaphroditism, primary hypogonadism and low-renin arterial hypertension with hypokalemia.
In the USSR, on the basis of the Institute of Experimental Endocrinology and Hormone Chemistry of the USSR Academy of Medical Sciences (later ENC), a children's endocrinology clinic was organized in 1961 under the leadership of prof. M.A. Zhukovsky. Clinical and scientific work on adrenogenital syndrome was actively carried out in the children's clinic [2, 15, 17, 18]. In 1969, the III All-Union Congress of Pediatric Doctors took place, at which topical issues of pediatric endocrinology were voiced [16, 19]. As a result of this congress, pediatric endocrinology was identified as a separate specialty. Already in June 1970, the First All-Union Conference of Pediatric Endocrinologists was held in Ivanovo, at which the most pressing problems of diagnosis and treatment of endocrine diseases in children were discussed; several reports were devoted to congenital dysfunction of the adrenal cortex [20].
In 1977, a simple and reliable method for screening for 21-hydroxylase deficiency, based on radioimmunoassay testing of 17-hydroxyprogesterone, became available for the first time [21]. The first pilot project of neonatal screening was carried out in Alaska in the Eskimo population, which has a high incidence of this pathology. According to the results of this project, the incidence of 21-hydroxylase deficiency among Alaskan Eskimos was 1:280 newborns and currently remains the highest incidence of this pathology in the world [22]. The introduction of neonatal screening made it possible to establish a diagnosis and begin treatment of the patient before the onset of a salt-wasting crisis, which significantly reduced mortality in this disease. In addition, screening has made it possible to diagnose the virile form of the disease in boys during the neonatal period, which significantly improves the growth prognosis of these patients. And finally, in girls with severe genital virilization at birth, thanks to the screening procedure, there are no errors in choosing their gender. By 1991, neonatal screening for adrenogenital syndrome was carried out in 13 countries around the world [23]. In Russia, the screening procedure has been introduced since 2006. The implementation of the project was carried out by the Federal State Institution Endocrinological Research Center (director - Academician I.I. Dedov), as the main endocrinological institution of the Ministry of Health and Social Development of the Russian Federation.
The widespread introduction of neonatal screening has opened up new opportunities for studying the epidemiology of CAH. By the middle of the 20th century in the United States, the prevalence of adrenogenital syndrome was first estimated as 1 case per 67,000 newborns [24]. However, the vast majority of patients had the viril or non-classical form of 21-hydroxylase deficiency, since without treatment, patients with the salt-wasting form died before diagnosis. Currently, the incidence of only classical forms of 21-hydroxylase deficiency, calculated based on neonatal screening data in the world population, is 1 case per 14,199 newborns [23]. Moreover, about 75% of cases occur in the salt-losing form and 25% in the simple virile form. The prevalence of the non-classical form of 21-hydroxylase deficiency is even higher, reaching 1 case per 1000 in the European population.
In the 1980s a new era began in the study of adrenogenital syndrome, associated with the successes of molecular genetics. Genes encoding enzymes of steroidogenesis were cloned, and a molecular diagnostic technique was developed for all forms of CDCN [25–28]. The absence of mutations in the CYP11A1 gene in patients with lipoid adrenal hyperplasia was unexpected. Further studies have shown that in most cases, lipoid adrenal hyperplasia is caused by a defect in the STAR protein, which ensures the transport of cholesterol to the inner mitochondrial membrane, where it becomes available to the action of the enzyme 11a-hydroxylase [29].
The development of molecular genetic methods for examining patients has made it possible to carry out prenatal diagnosis of adrenogenital syndrome. Soon, French and American scientists proposed prenatal therapy with dexamethasone, the purpose of which was to prevent intrauterine virilization of female fetuses with deficiency of 21-hydroxylase and 11b-hydroxylase [30, 31]. Dexamethasone is not affected by the enzyme 11b-hydroxysteroid dehydrogenase type 2 in the placenta, therefore, it enters the fetus and, through a negative feedback mechanism, reduces the level of ACTH, thereby preventing excessive secretion of adrenal androgens, and prevents virilization of the external genitalia in a female fetus.
In 2004, a group of British scientists discovered another new form of CDCN—oxidoreductase deficiency caused by a defect in the POR gene [32]. Patients with a clinical picture of combined deficiency of P450C17 and P450C21 were described back in 1985, but no mutations were found in the genes of these enzymes, and no one could explain the mechanism for the development of such manifestations until 2004. Cytochrome P450 oxidoreductase provides electrons to all microsomal P450 enzymes, including CYP51A1, CYP17A1, CYP21A2, and CYP19A1, so the clinical picture reflects a combined deficiency of these enzymes. Relative deficiency of 21-hydroxylase (CYP21A2) leads to intrauterine virilization of fetuses with karyotype 46XX; deficiency of 17a-hydroxylase (CYP17A1) can lead to insufficient masculinization of fetuses with karyotype 46XY and to the development of androgen deficiency during puberty; CYP51A1 deficiency can lead to the formation of bone abnormalities such as craniosynostosis, radial-ulnar synostosis and multiple contractures; Aromatase deficiency (CYP19A1) can cause hypogonadism in women, and placental aromatase deficiency leads to maternal virilization during pregnancy. The description of oxidoreductase deficiency proves that it is too early to put an end to the description of the mechanisms of development of disorders of steroidogenesis in the adrenal glands.
In the last decade, the main efforts of doctors have been aimed at optimizing the treatment of adrenogenital syndrome; long-term follow-up observation has revealed complications and shortcomings of standard treatment regimens. In addition to glucocorticoid therapy, the use of growth hormone drugs in combination with long-acting luliberin analogues has been proposed to improve the growth prognosis of patients [33]. New glucocorticoid drugs with altered pharmacodynamic characteristics are at the stage of clinical testing [34].
The data presented describe the evolution of our ideas about the mechanisms of the occurrence of CAH and methods of treating patients with this pathology of the adrenal glands. In the past, a rare, difficult to recognize disease became the object of population screening in many countries, including Russia. Of course, in the near future, many organizational problems will have to be solved related to screening and providing qualified care to patients living in regions of the country remote from large centers. Our nearest “tomorrow,” with the help of a modern healthcare organization and highly qualified endocrinologists, should rid society of erroneous or missed diagnoses of CAH in newborns and ensure these patients full physical and psychological development and the possibility of reproduction.
But besides these administrative and economic tasks, which are undoubtedly of national importance for Russia, there remain many questions that scientists and doctors around the world have to solve. It should be noted that a certain category of patients still does not fully meet the diagnostic criteria for the currently known variants of steroidogenesis defects in the adrenal glands. This is confirmed by the discovery of a defect in oxidoreductase. And this new form, which was recently isolated in the VDKN group, is probably not the last. Clinicians, describing and publishing unusual observations, gradually form hypotheses about new, not yet studied enzymes and coenzymes of steroidogenesis, which can lead to similar disorders. Questions about treatment regimens, adaptation of patients to male or female gender in different age groups, issues of reproduction, prenatal diagnosis and treatment - all these topics are still the subject of heated debate, and new research provides new arguments.
If we talk about the future, perhaps even about the day after tomorrow, we cannot ignore the topic that has been exciting the minds of scientists and doctors for decades - “gene therapy.” Genetics is the science of the present and future, on which hopes are pinned in the treatment of many diseases. The first publication devoted to gene therapy for hereditary diseases was published in 1972 in the journal Science (“Gene therapy for human genetic disease?”) [35]. In 1990, at the US National Institutes of Health, this method was first used to treat congenital immunodeficiency in humans (a temporary effect was observed). But since then, the method has not become publicly available and widely used, since its application is associated with many technical and ethical difficulties. Defects in the genes for steroidogenesis enzymes have not yet become the object of gene therapy, but the method of prenatal and preimplantation diagnosis of CDCN has already been widely implemented, which can significantly increase the likelihood of having a healthy child using in vitro fertilization methods [36].
Conclusion
CHD is one of many hereditary diseases that is no longer classified as “rare”. Everyone recognizes the enormous social significance of this problem in healthcare. Significant advances in world science give us the opportunity to make people with this pathology normal members of society, capable of learning, working and bearing children. In Russia, the diagnosis and treatment of CHD is carried out at a modern level, there is extensive experience in the largest endocrinological centers in the country, and newborn screening has been introduced. The immediate priority task of the endocrinological community in Russia is to provide modern, qualified care to all patients. The role of scientific research in this area in Russia, both clinical and experimental, is also great, which will contribute to world endocrinology.
Causes
The causes of adrenogenital syndrome are associated with a genetic disorder - a defect in one of the genes of the 6-1 pair of chromosomes. This gene is recessive and only appears if a child is born with both altered chromosomes. His parents must either be carriers of the pathological gene or suffer from this disease.
This gene is responsible for the synthesis of the enzyme 21-hydroxylase. Its deficiency or absence leads to compensatory increased synthesis of the pituitary hormone ACTH, which leads to adrenal hypertrophy and increased production of androgens. They suppress ovarian function and the formation of female sex hormones - estrogens.
Why this gene mutates is unknown. Both boys and girls can be carriers of the altered gene with equal frequency. The clinical development of the disease also does not depend on the sex of the child, or on which parent is sick and who only passed on the pathological gene.
2. Reasons
As stated above, the direct cause of the development of adrenogenital syndrome is a deficiency of the 21-hydroxylase protein caused by a genetic mutation. There are three main forms:
- with salt-wasting syndrome (complete absence of the enzyme);
- simple viril form (partial deficiency of 21-hydroxylase);
- non-classical postpubertal form.
The first option is the most common; its share in the total number of cases of adrenogenital syndrome reaches 75%.
The acquired postpubertal form, which develops already in adulthood, is most often caused by an oncological process in the adrenal cortex.
Visit our Endocrinology page
Symptoms and diagnosis of the disease
Patients should be observed by a pediatric endocrinologist.
Competent treatment from this specialist makes it possible to normalize hormonal function. Symptoms of adrenogenital syndrome:
- rapid growth during adolescence;
- the occurrence of hirsutism in girls - hair on the upper lip, inner thighs, along the white line of the abdomen, around the nipples;
- severe acne;
- menstrual irregularities, infertility.
External signs of adrenogenital syndrome are most noticeable in girls:
- male body type - tall, narrow pelvis, underdeveloped mammary glands, broad shoulders; weight within normal limits;
- violation of the structure of the external genitalia with a normally formed uterus and a woman’s natural sexual orientation;
- When pregnancy occurs, a frequent complication is termination in the 1st trimester.
Diagnosis of adrenogenital syndrome is carried out using the following studies:
- determination of the level of hormones: testosterone, DHEA, DHEA-S, 17-OHP, LH, FSH, showing the degree of dysfunction of the adrenal glands and ovaries;
- tests with dexamethasone and ACTH, necessary for differential diagnosis with polycystic ovary syndrome;
- Ultrasound of the ovaries;
- determining the duration of cycle phases by charting basal temperature.
Diagnosis of adrenogenital syndrome must be entrusted to a qualified specialist. A prerequisite is the presence of a modern laboratory for hormonal research. The Mama Papa Ya clinic offers all these services to its patients.
What will happen if AGS is not treated?
An excess of androgens leads to improper development of the genital organs in girls. Disorders range from mild enlargement of the clitoris to pronounced signs characteristic of men. In severe cases, it is difficult to determine the sex of the baby because the clitoris is similar to the penis and the urethral opening is located on the head.
The abnormal development of the labia majora gives them a resemblance to the scrotum. In this disease, the internal genital organs are developed according to the female type, but sometimes the sex of the child can only be determined by ultrasound.
Later, girls develop other male characteristics:
- low voice;
- muscular build;
- male-pattern hair growth, much faster than age-related. By the age of four, hair begins to appear under the armpits, and at 8-9 years old - on the face.
By the beginning of adolescence, girls have a specific physique:
- short stature caused by rapid closure of bone growth zones;
- large body with short limbs;
- the shoulder region is wider than the pelvic region;
- undeveloped mammary glands.
In mild forms of adrenogenital syndrome, there are menstruation, but they begin later, are painful (algomenorrhea) and are quite scanty. In severe forms, menstruation is completely absent.
If AGS is not treated, a woman will have serious problems for the rest of her life: she will have masculine features, but an internal feminine structure. However, she will never become a mother.
Treatment
In the salt-wasting form of the disease, treatment begins immediately after the birth of the child, as soon as the first symptoms appear. It includes intensive therapy with the administration of fluids, salts, and hormonal drugs.
Treatment of adrenogenital syndrome includes:
- long-term use of adrenal hormones - glucocorticoids, which should begin in early childhood (up to 7 years);
- according to indications - prescription of female sex hormones, especially in preparation for pregnancy;
- If necessary, even in childhood, patients undergo surgical plastic surgery of the external genitalia.
Maintaining pregnancy in sick women causes significant difficulty. However, a competent gynecologist-endocrinologist is able to select the necessary medications so that the patient can give birth to a healthy child.
Symptoms of adrenal cortex dysfunction
With the congenital variant of adrenogenital syndrome, the figure of patients already in childhood acquires masculine features. Their growth zones in the bones close early, so they are short in stature, their voices become rougher, and their genitals have an irregular structure (hypertrophied clitoris, modified labia resembling a scrotum).
In addition to the classic form of AGS, there are:
- pubertal, when symptoms appear in adolescence, during the period of physiological enhancement of the hormonal function of the adrenal cortex;
- postpubertal, when manifestations manifest at the end of the second decade of life, often after a spontaneous miscarriage in early pregnancy, a non-developing pregnancy or after a medical abortion;
- latent form, the only symptom of which may be miscarriage in the early stages.
When studying the karyotype, 46 XX chromosomes are determined (female set). The internal genital organs also correspond to the female body (uterus, ovaries). Puberty occurs early (at 6-7 years): hair growth develops according to the male type, the mammary glands are poorly developed, menstruation is often absent, however, a full menstrual cycle may be present. In the future, patients with this diagnosis will experience infertility or recurrent miscarriage.
If the dysfunction of the adrenal cortex is secondary, patients complain of:
- sudden appearance of excess hair (hirsutism);
- irregular menstrual cycle;
- inability to get pregnant;
- reduction of mammary glands;
- increasing the size of the clitoris.
When performing an ultrasound of internal organs, underdevelopment or atrophy of the uterus and ovaries is diagnosed.
In both cases, the pathology can develop with a predominance of one of the syndromes:
- Salt-wasting: patients note general virilization (masculinity), hypotension (low blood pressure), edema.
- Hypertensive: manifested by damage to the myocardium and blood vessels, increased blood pressure.
- Virilization: the figure and voice of patients become more masculine, but there is no damage to the internal organs.
Make an appointment
Why you need to contact the Mama Papa Ya clinic
The network of family clinics “Mama Papa Ya” is located in Moscow and other cities. For adrenogenital syndrome, the following medical services are provided at an affordable price:
- laboratory diagnosis of hormonal disorders using modern equipment;
- reception of specialized specialists - pediatrician, gynecologist, endocrinologist;
- consistent monitoring of the child, continuity in treatment by doctors of different profiles;
- modern methods of treatment during pregnancy;
- prescription of drug therapy using modern methods.
Doctors at our clinic carry out early diagnosis and prescribe competent treatment for any form of the disease. Under such conditions, the manifestations of adrenogenital syndrome are well corrected. Make an appointment right now!
Reviews
Good clinic, good doctor!
Raisa Vasilievna can clearly and clearly explain what the problem is. If something is wrong, she speaks about everything directly, not in a veiled way, as other doctors sometimes do. I don’t regret that I ended up with her. Anna
I would like to express my gratitude to the staff of the clinic: Mom, Dad, and me. The clinic has a very friendly atmosphere, a very friendly and cheerful team and highly qualified specialists. Thank you very much! I wish your clinic prosperity.
Anonymous user
Today I had a mole removed on my face from dermatologist I.A. Kodareva. The doctor is very neat! Correct! Thanks a lot! Administrator Yulia Borshchevskaya is friendly and accurately fulfills her duties.
Belova E.M.
Today I was treated at the clinic, I was satisfied with the staff, as well as the gynecologist. Everyone treats patients with respect and attention. Many thanks to them and continued prosperity.
Anonymous user
The Mama Papa Ya clinic in Lyubertsy is very good. The team is friendly and responsive. I recommend this clinic to all my friends. Thanks to all doctors and administrators. I wish the clinic prosperity and many adequate clients.
Iratyev V.V.
We visited the “Mama Papa Ya” Clinic with our child. A consultation with a pediatric cardiologist was needed. I liked the clinic. Good service, doctors. There was no queue, everything was the same price.
Evgeniya
I liked the first visit. They examined me carefully, prescribed additional examinations, and gave me good recommendations. I will continue treatment further; I liked the conditions at the clinic.
Christina
The doctor carefully examined my husband, prescribed an ECG and made a preliminary diagnosis. She gave recommendations on our situation and ordered additional examination. No comments so far. Financial agreements have been met.
Marina Petrovna
I really liked the clinic. Helpful staff. I had an appointment with gynecologist E.A. Mikhailova. I was satisfied, there are more such doctors. Thank you!!!
Olga