Организм человека состоит из огромного количества клеток. Они в свою очередь объединяются в более крупные единицы – ткани и органы. Из последних строятся системы органов, а и из них и получается организм. И на всех уровнях постоянно идут различные взаимодействия и сложные биохимические реакции. Чтобы все это гармонично развивалось из оплодотворенной яйцеклетки и слаженно функционировало, требуется специальная схема работы для всех уровней. Ее роль в организме выполняет организованная генетическая информация.
Именно на наследственный материал возлагается задача по сохранению данных об общем облике человека, особенностях строения отдельных органов, принципах регуляции при помощи гормонов, и даже того, каким образом должны собираться белки. Все данные записываются в виде последовательности структурных единиц – нуклеотидов. Они подобно буквам алфавита формируют группы-слова, выполняющие определенные функции. Каждая группа называется геном. Все вместе они формируют молекулу ДНК. Если посмотреть на уровень выше, то можно заметить, что весь наследственный материал в виде цепочек дезоксирибонуклеиновой кислоты сконцентрирован в особых образованиях, которые называются хромосомами.
Их у человека в норме насчитывается 23 пары. В каждой паре информация хранится в двух копиях. Это необходимо для того, чтобы при передаче генетической информации каждая дочерняя клетка получила свою копию. Одна пара хромосом отличается от остальных и отвечает за вопросы определения пола. Если в ней имеются две одинаковые большие хромосомы (ХХ вариант), то организм относится к женскому полу. Если же имеется одна большая и одна малая хромосома, то речь идет о мужском поле (XY вариант). Эти две отличающиеся от прочих хромосомы называются половыми. Оставшиеся 22 пары встречаются во всех организмах независимо от пола. Их называют аутосомами.
Аномалии генетического материала
Наследственный материал состоит из огромного количества нуклеотидов, формирующих гены. При этом в каждом гене последовательность нуклеотидов строго определена, поскольку должна кодировать определенный белок. Кроме того, сами гены при формировании хромосом также выстраиваются в фиксированном порядке. Благодаря сохранению этого порядка организм может функционировать, а ученые – быстро и точно указывать друг другу, про какой ген идет речь.
В идеальном случае система работает без малейших сбоев, а генетическая информация всегда передается в неизменном виде. Однако на практике большое число структурных единиц и постоянное воздействие различных факторов (например, ионизирующего излучения) приводит к тому, что время от времени возникают различные аномалии. В частности, отдельные участки последовательности ДНК могут быть скопированы на новое место. В таком случае говорят о дупликации. Если же вместо создания новой копии была перемещена часть исходной цепочки, то модификация называется транслокацией. Кроме того, иногда часть последовательности просто теряется, удаляется из генетического материала. В таком случае изменение называется делецией.
Поскольку взаимодействия в организме оттачивались в течение многих тысячелетий эволюционного развития, получилась очень слаженная система. И аномалии, даже самые небольшие, могут вызвать нарушение баланса. В таком случае в организме развивается то или иное нарушение. Если при этом причина находится на уровне генов, то говорят о генных болезнях. Если была утрачена или наоборот получена лишняя копия хромосомы, то такие нарушения называются хромосомными заболеваниями.
Симптомы и признаки
Клиническая картина недуга весьма разнообразна и может отличаться в каждом конкретном случае. В частности, у ребенка проявляются физические и психические отклонения, некоторые признаки характерны для всех случаев, другие встречаются значительно реже.
| Физические отклонения | Психические отклонения |
|
|
Клинические проявления недуга принято делить также в зависимости от их распространенности. Так, существуют симптомы, которые встречаются у всех детей, страдающих данным недугом, признаки, возникающие в большинстве случаев (но не всегда) и более редкие проявления.
| Характерные симптомы | Частые признаки | Более редкие проявления |
|
|
|
Клинические проявления недуга меняются по мере взросления ребенка. Так, у детей подросткового возраста наблюдается задержка полового созревания (при этом детородная функция в дальнейшем не нарушается), а когда ребенок становится взрослым, он выглядит значительно моложе своих сверстников.
Нередко у детей старшего возраста возникают и другие проявления заболевания, такие как нарушение мочеиспускания, сложные нарушения моторики рук, проблемы с общением (подросток понимает устную речь, но не видит необходимости отвечать собеседнику).
Какова продолжительность жизни детей с синдромом Вильямса? Ответ узнайте прямо сейчас.
Что такое микроделеционный синдром?
Самые незначительные изменения (они же мутации) называются точечными. Их появление влияет на считанные единицы генов. В некоторых случаях нарушение относится вообще к одному единственному гену. Однако если он обеспечивал выработку важного белка, последствия для всего организма могут быть очень серьезными. Подобные патологические изменения относятся к группе микроделеционных синдромов.
Каждое такое заболевание обусловлено небольшим изменением генетического материала, которое происходит в строго определенном месте. Точный механизм возникновения подобных нарушений на сегодняшний день не установлен, что не мешает ученым заниматься исследованием их воздействия на организм.
Так, было выяснено, что развитие синдрома в таком случае может происходить несколькими различными вариантами. В частности, ряд заболеваний характеризуется участием онкогенов. В других случаях на воздействие непосредственно самой делеции накладывается эффект хромосомного импринтинга и возможные однородительские дисомии.
Частота возникновения большей части микроделеционных синдромов крайне невелика: порядка 1 случая на 50-100 тысяч новорожденных. Набор клинических признаков обычно выражается отчетливо. Для того чтобы поставить диагноз, бывает достаточно лишь совокупности симптомов. Однако при таком подходе невозможно точно прогнозировать здоровье потомков, поэтому зачастую наряду с проверкой обычных признаков производится молекулярно-генетическая диагностика пробанда и его родственников (обычно родители, в некоторых случаях также требуется анализ генотипа братьев, сестер, теть, дядь и так далее).
Патологические проявления сильно отличаются. В частности, их проявление определяется тем, насколько большой участок генетического материала был утрачен в результате делеции. Кроме того, в ряде случаев играет роль то, от кого из родителей была получена мутация (влияние хромосомного импринтинга). Хорошей иллюстрацией последней ситуации является пара синдромов Прадера-Вилли и Ангельмана. Они оба обусловлены наличием делеции в 15 хромосоме. Однако из-за различного механизма действия при передаче от разных родителей клиническая картина этих заболеваний значительно отличается.
История [ править ]
«Мальчик с куклой» или «Ребенок с рисунком», около 1520 года, Джованни Франческо Карото ; портрет возможного случая синдрома Ангельмана. [35]
Гарри Ангелман , педиатр, работающий в Уоррингтоне , Англия, впервые сообщил о трех детях с этим заболеванием в 1965 году. [10] Ангелман позже описал свой выбор названия «Дети-марионетки», чтобы описать эти случаи как связанные с картиной маслом, которую он видел во время отпуска в Италии:
История медицины полна интересных историй об открытии болезней. Сага о синдроме Ангельмана — одна из таких историй. Совершенно случайно почти тридцать лет назад (например, примерно в 1964 году) трое детей-инвалидов были приняты в разное время в мое детское отделение в Англии. У них были разные недуги, и хотя на первый взгляд казалось, что они страдают от разных заболеваний, я чувствовал, что у их болезни была общая причина. Диагноз был чисто клиническим, потому что, несмотря на технические исследования, которые сегодня стали более изощренными, мне не удалось установить научное доказательство того, что у всех троих детей был одинаковый дефект. В связи с этим я не решался писать о них в медицинских журналах. Однако во время отпуска в Италии мне довелось увидеть картину маслом вМузей Кастельвеккьо в Вероне назвал … Мальчик с куклой. Смеющееся лицо мальчика и тот факт, что мои пациенты судорожно двигались, натолкнули меня на мысль написать статью о трех детях под названием «Дети-марионетки». Это имя нравилось не всем родителям, но оно служило средством объединения трех маленьких пациентов в единую группу. Позже название было изменено на синдром Ангельмана. Эта статья была опубликована в 1965 году и после некоторого первоначального интереса оставалась почти забытой до начала восьмидесятых.
- Ангелман, цитата Чарльза Уильямса [36]
Сообщения о случаях заболевания из США впервые начали появляться в медицинской литературе в начале 1980-х годов. [37] [38] В 1987 году было впервые отмечено, что примерно у половины детей с AS отсутствует небольшой кусочек хромосомы 15 ( частичная делеция хромосомы 15q
). [39]
Позитивное влияние некоторых делеций на жизнеспособность
Небольшие изменения делеционного характера могут существенно повлиять на выживание организма. К примеру, утрата гена, кодирующего белок CCR5-δ32, становится причиной невосприимчивости к вирусу иммунодефицита человека. Ученые предполагают, что эта мутация впервые появилась около 2,5 тысячелетий назад и с течением времени распространилась по территории Европы.
Имеющееся на сегодняшний день распределение отличается неравномерностью. Согласно статистическим данным, около 10% жителей европейских стран устойчиво к ВИЧ. Вместе с тем в скандинавских государствах этот показатель достигает 14-15 процентов. Русские и финны демонстрируют 16-процентный уровень устойчивости. В то же время для Сардинии частота равняется скромным 4 процентам.
Ряд ученых выдвинул гипотезу, что подобное распространение определяется прошедшими в средневековый период эпидемиями бубонной чумы. Вероятно, мутация в гене вызывает повышенную сопротивляемость этому заболеванию. Поэтому на территории тех стран, где прошлась «черная смерть», выжило больше людей с этим генотипом.
Влияние делеций на способность к оплодотворению
Делеции, происходящие в обычных хромосомах (аутосомах) могут в некоторых случаях быть компенсированы нормальной копией гена. Однако когда речь заходит о половых хромосомах, особенно об Y-хромосоме, ситуация меняется.
Прежде всего необходимо отметить, что локализованные на ней гены не имеют второго экземпляра. При нормальном количестве хромосом в наборе Y-хромосома оказывается крайне уязвимой. В сочетании с малым количеством генов это приводит к серьезным последствиям каждого изменения. Особый интерес представляют мутации, касающиеся AZF-локуса и SRY гена.
Аномалии гена SRY (Sex-determining Region Y)
Ген SRY, как следует из его названия, отвечает за крайне важную функцию. Именно его наличие в хромосомном наборе запускает процесс формирования организма по мужскому фенотипу и стимулирует развитие соответствующих половых органов.
Наличие даже небольшой делеции в этом гене нарушает механизм дифференцировки пола. В результате при нормальном кариотипе 46XY зародыш начинает развиваться как женский организм. По этой причине на ген SRY приходится наибольшее число мутаций, связанных с неразвитостью гонад. Кроме того, изменения этого гена вызывают инверсию пола.
Аномалии AZF-локуса
На Y-хромосоме также имеется особый участок, который контролирует процесс выработки сперматозоидов. Именно от этого зависит, насколько эффективным будет сперматогенез. Кроме того, состояние этого участка сказывается на свойствах сперматозоидов, таких как общее количество в эякуляте, способность двигаться, наличие структурных изменений и способность к оплодотворению. Только при наличии хорошо сформированных подвижных сперматозоидов мужской генетический материал может быть доставлен до яйцеклетки. Иными словами, о состоянии этого небольшого участка генетического кода зависит способность мужчины иметь детей.
При наличии в AZF-локусе аномалий процесс выработки сперматозоидов нарушается. В результате могут развиться азооспермия и олигозооспермия. При этих патологиях в эякуляте либо совсем не содержится сперматозоидов, либо их число сильно снижено.
Сам AZF локус делится на три части со специфическими задачами. Они именуются путем добавления индекса: AZFa, AZFb и AZFc. Возникшая делеция может удалять фрагмент отдельной части, либо ее целиком, либо захватывать сразу два региона. При полном удалении AZF развивается тяжелое поражение сперматогенеза. Частичные делеции могут проявляться по-разному. При этом на степень проявления патологии влияют размеры утраченного фрагмента и его расположение в локусе. Поэтому для прогностических целей крайне важно знать, в каком месте произошла делеция. Кроме того, эта информация может использоваться для правильного планирования семьи и проведения экстракорпорального оплодотворения.
Если при делеции был удален весь локус или любой из регионов с индексами a/b, то у мужчины не могут быть получены жизнеспособные сперматозоиды. Если делецию можно описать формулой AZFb/AZFb+, то развивается азооспермия из-за тяжелых нарушений процесса формирования сперматозоидов.
Делеции участка AZFc приводят к проявлению патологических симптомов различной степени тяжести. В том числе возможно развитие олигоспермии, которая в принципе допускает зачатие. В 50-70 процентах от общего числа подобных случаев возможно получение сперматозоидов для дальнейшего использования в методах искусственного оплодотворения. Частичная делеция региона AZFс может выражаться в форме различных нарушений от нормозооспермии до азооспермии.
Все делеции в AZF-локусе, вызывающие ту или иную патологическую ситуацию, являются причинами мужского бесплодия. Определение мутации возможно путем гистологического анализа семенной жидкости. При этом необходима остановка созревания сперматозоидов или обнаружение незрелых сперматозоидов. Для получения точных данных о делециях в AZF-локусе используется ПЦР 6 маркеров, которые относятся к отдельным участкам локуса.
Причины и провоцирующие факторы
Основной причиной развития недуга считается изменение или отсутствие одного из фрагментов 15 хромосомы. Риск развития патологии у ребенка повышается, если у его близких родственников (родителей) имеются следующие генетические отклонения:
- Присутствие лишних хромосом (одной или нескольких).
- Хромосомная инверсия, когда один из участков хромосомы перевернут, при этом часть его фрагмента утеряна, а гены располагаются в противоположном порядке.
- Перестройка Y-хромосомы, когда 2 ее участка меняются местами, при этом некоторые фрагменты мутировавшей хромосомы могут отсутствовать.
- Утрата одного или нескольких хромосомных фрагментов.
- Присоединение фрагмента одной хромосомы к другой.
- Наличие лишних генов, возникших в результате репликации одной или нескольких хромосом.
- Кольцевое строение хромосомы, когда концы ее соединяются между собой, образуя кольцо.
Синдром Ангельмана
При синдроме Ангельмана развивается характерный набор патологических изменений. В частности, отмечается задержка психологического развития, сопровождающаяся проблемами со сном, частыми хаотическими движениям (больше руками), постоянными улыбками и смехом.
Патология развивается при отсутствии некоторых генов, расположенных на 15 хромосоме. При этом обязательным условием является передача мутантной копии гена от матери. Если поврежденная хромосома будет унаследована от отца, то разовьется синдром Прадера-Вилли. Кариотип обычно нормальный (46XX и 46XY для девочек и мальчиков соответственно). Различные независимые исследования указывают на связь болезни с геном UBE3A, который в норме обеспечивает выработку ферментного компонента в сложной системе деградации белков.
Частота появление синдрома составляет примерно 1 случай на 10-20 тысяч новорожденных (показатели отличаются у различных ученых).
Характерными особенностями больных с синдромом Ангельмана являются следующие признаки:
· проблемы с питанием, начинающиеся еще во время грудного вскармливания, поскольку дети плохо набирают вес (распространенность признака порядка 75 процентов);
· заторможенное развитие навыков общей моторики, то есть дети позже других начинают сидеть и ходить;
· для всех детей характерны нарушения речевого развития;
· больные обычно понимают больше, чем в состоянии выразить при помощи ограниченного словарного запаса;
· часто заболевание сопровождается дефицитом внимания и гиперактивностью;
· проблемы с обучением в обычной школе;
· у 80% заболевших развивается эпилепсия, сопровождающаяся заметными на электроэнцефалографии нарушениями; ученые полагают, что заболевание эпилепсией носит вторичный (симптоматический) характер.
· выполнение необычных движений, к которым относятся произвольные хаотические движения конечностями, мелкий тремор;
· возникновение приступов смеха при отсутствии видимых причин;
· характерная ходьба на негнущихся ногах, из-за которой возникло сравнение с марионетками;
· уменьшенная по сравнению со средними размерами голова, часто с уплощенным затылком;
· в некоторых случаях встречаются своеобразные запоминающиеся черты лица – широкий рот с редко расположенными зубами, выдвинутый вперед подбородок с выпущенным наружу языком;
· различные нарушения сна;
· примерно в 40 процентах случаев развивается косоглазие;
· порядка 10% больных также страдает от искривления позвоночника;
· высокие температуры воспринимаются с повышенной чувствительностью;
· наибольшего комфорта обычно достигают в воде (к примеру, в ванной)
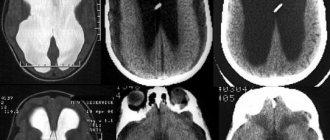
Как правило, синдром определяется при помощи методов молекулярно-генетической диагностики по 15 хромосоме. Показанием к проведению тестирования для новорожденного является пониженный мышечный тонус (гипотонус), заметное отставание в развитии речи и мелкой моторики. Кроме того, на заболевание могут указывать мелкий тремор, порывистые беспорядочные движения, передвижение на негнущихся ногах.
Анализ может проводиться через флуоресцентную гибридизацию in situ, метилированием ДНК в области 15q11-q13. Также можно проверить мутации в импринтинговом центре и в гене UBE3A.
Поскольку заболевание обусловлено генетическим нарушением, адекватного и действенного способа лечения для него не имеется. Выполнение лечебных мероприятий, таких, как массаж для больных с гипотонусом, позволяет повысить качество жизни.
Уход за ребенком и прогноз заболевания
Болезнь обнаруживают в основном, когда ребенок находится в возрасте от трех до семи лет. Симптоматика выражена четко именно к достижению этого возраста, не раньше. Поскольку симптомы неспецифичны, тяжело диагностировать рассматриваемый синдром у младенца. Важно отметить, что болезнь относительно редкая. Потому врач, есть вероятность, не сможет и догадаться, что он имеет дело с синдромом Ангельмана.
В Европе есть больше данных про рассматриваемую патологию, потому диагностика у них более качественная и быстрая. Поставить диагноз должен врач-генетик, который назначает генетический анализ, на основе которого делаются выводы.
Прогноз болезни коррелирует со степенью поражения пятнадцатой хромосомы. Часть пациентов нормально ведут себя в быту, общение у них также почти нормальное. Но в части случаев ребенок не может ходить, речь у него ответствует. Люди с синдромом Ангельмана живут 20-50 лет. Качество жизни во многом зависит от тяжести синдрома, как уже было сказано выше.
Для жизни больного важно понимание и внимание его семьи. Они должны понять ребенка и дать ему всю любовь, какую только могут, чтобы его психическое развития было полноценным. Часть детей приходится отдавать в интернат или специализированную школу. Там они обучаются по специальным программам и учатся жить в обществе.
Синдром Прадера-Вилли
Это заболевание определяется той же самой генетической мутацией, что и для синдрома Ангельмана. Отличие состоит в том, что при этом нарушение наследственного материала получается со стороны отца. Кариотип соответствует нормальному (46XX или 46XY). По распространенности (1 случай на 12-15 тысяч новорожденных) примерно совпадает с распространенностью синдрома Ангельмана.
Характерными признаками синдрома Прадера-Вилли являются следующие симптомы:
· в пренатальный период малая подвижность плода;
· часто встречается неверное положение плода;
· возможна дисплазия тазобедреных суставов;
· к двум годам может проявиться склонность много есть (больше нормы), что приводит к ожирению;
· низкий мышечный тонус (гипотонус), сочетающийся с нарушенной координацией движений;
· стопы и кисти обычно маленькие, кроме того характерен невысокий рост;
· формирование косоглазия и сколиоза;
· отмечают повышенную сонливость;
· плотность костей находится на более низком уровне, чем у здоровых людей;
· слюна густая, обычно состояние зубов плохое;
· недостаточная функция половых желез, вызывающая в итоге бесплодие;
· позднее по сравнению со сверстниками половое созревание;
· больные позже учатся говорить, отстают в психическом развитии;
· внешние признаки включают выраженную переносицу, узкий и высокий лоб, миндалевидную форму глаз, узкие губы.
В большинстве случаев у человека с мутацией насчитывается от одного до пяти признаков заболевания.
Диагностика заболевания проводится путем молекулярно-генетического тестирования, на которое направляются дети с пониженным мышечным тонусом. Зачастую вместо верного диагноза определяется более распространенный «синдром Дауна». Опытный генетик, достаточно часто встречающийся с проявлениями синдрома Прадера-Вилли способен диагностировать его по комплексу внешних признаков.
Ссылки [ править ]
- ^ a b c Синдром Ангельмана , взято из Оксфордского словаря английского языка Lexico.com
- ^ a b c d Синдром Ангельмана в Медицинском словаре Merriam-Webster.com
- Зима, Робин М .; Барайцер, Майкл (2013). Множественные врожденные аномалии: диагностический сборник . Springer. п. 34. ISBN 9781489931092. Архивировано 5 ноября 2021 года.
- Синдром Ангельмана в Словаре научных и технических терминов Макгроу-Хилла
- ^ a b Синдром Ангельмана в Медицинском словаре американского наследия
- Синдром Ангельмана в комплексной нейробиологии развития: развитие и функции нервной цепи в здоровом и больном мозге: Глава 32.
- ^ a b c d e f g h i j k l m n o p Ссылка, Genetics Home (май 2015 г.). «Синдром Ангельмана» . Домашний справочник по генетике
. Архивировано 27 августа 2021 года . Проверено 28 апреля 2017 года . - ^ a b c d e f g h i j k «Синдром Ангельмана — NORD (Национальная организация по редким заболеваниям)» . NORD (Национальная организация по редким заболеваниям)
. 2015. Архивировано 13 ноября 2021 года . Проверено 28 апреля 2017 года . - ^ a b «Распространенные неправильные диагнозы | БЫСТРО» . FAST (Фонд терапии синдрома Ангельмана)
. Проверено 10 октября 2021 . - ^ a b Ангелман, Гарри (1965). « Дети — марионетки: отчет о трех случаях». Dev Med Child Neurol
.
7
(6): 681–688. DOI : 10.1111 / j.1469-8749.1965.tb07844.x , S2CID 53730099 . - Уилсон, Голдер Н .; Кули, В. Карл (2000). Профилактическое ведение детей с врожденными аномалиями и синдромами . Издательство Кембриджского университета. п. 193. ISBN. 9780521776738. Архивировано 5 ноября 2021 года.
- Кумар, Винай; Аббас, Абул К .; Астер, Джон С. (2013). Базовая патология Роббинса . Elsevier Health Sciences. п. 244. ISBN 978-1437717815. Архивировано 5 ноября 2021 года.
- ↑Факты о синдроме Ангельмана (PDF). Архивировано 27 мая 2013 г. в Wayback Machine
. Анонимный. Сайт Фонда синдрома Ангельмана (США). Проверено 29 сентября 2012 года. - Weeber Е, Левенсон J, Sweatt J (2002). «Молекулярная генетика познания человека». Молекулярные вмешательства
.
2
(6): 376-91, 339. DOI : 10,1124 / mi.2.6.376 . PMID 14993414 . - Ван, Иян и др. (2017). «Идентификация целей убиквитинирования E6AP путем ортогонального переноса убиквитина» . Nature Communications
.
8
(1): 2232. Bibcode : 2017NatCo … 8.2232W . DOI : 10.1038 / s41467-017-01974-7 . PMC 5738348 . PMID 29263404 . - Mabb, AM; Джадсон, MC; Зилка, MJ; Филпот, Б. Д. (май 2011 г.). «Синдром Ангельмана: понимание геномного импринтинга и фенотипов развития нервной системы» . Trends Neurosci
.
34
(6): 293–303. DOI : 10.1016 / j.tins.2011.04.001 . PMC 3116240 . PMID 21592595 . - Кэссиди, SB; Дайкенс, Э (2000). «Синдромы Прадера-Вилли и Ангельмана: сестринские запечатленные расстройства». Am J Med Genet
.
97
(2): 136–146. DOI : 10.1002 / 1096-8628 (200022) 97: 2 <136 :: помощь-ajmg5> 3.0.co; 2-V . PMID 11180221 . - White HE, Durston VJ, Харви JF, Cross NC (2006). «Количественный анализ метилирования гена SNRPN (коррекция SRNPN) методом пиросеквенирования как диагностический тест для синдрома Прадера – Вилли и синдрома Ангельмана» . Clin.Chem
.
52
(6): 1005–13. DOI : 10,1373 / clinchem.2005.065086 . PMID 16574761 . - ^ a b c Williams C (2005) «Неврологические аспекты синдрома Ангельмана» Мозг и развитие 27: 88–94
- Лаан, Лаура AEM; Вена, Алла А. (2005). «Синдром Ангельмана: есть ли характерная ЭЭГ?». Мозг и развитие
.
27
(2): 80–87. DOI : 10.1016 / j.braindev.2003.09.013 . ISSN 0387-7604 . PMID 15668045 . Кириллович 5912 . - Дэн, Б., Синдром Ангельмана: текущее понимание и перспективы исследования. Эпилепсия, 2009. 50 (11): с. 2331–2339.
- Сидоров, Михаил С .; Колода, Джина М .; Долатшахи, Марджан; Thibert, Ronald L .; Bird, Lynne M .; Чу, Кэтрин Дж .; Филпот, Бенджамин Д. (8 мая 2021 г.). «Дельта-ритмичность — надежный биомаркер ЭЭГ при синдроме Ангельмана: параллельный анализ мыши и человека» . Журнал нарушений развития нервной системы
.
9
: 17. DOI : 10,1186 / s11689-017-9195-8 . ISSN 1866-1955 . PMC 5422949 . PMID 28503211 . - ^ a b c Фролих, Джоэл, Меган Миллер, Линн М. Берд, Пилар Гарсес, Ханна Пурелл, Мариус К. Хоэнер, Бенджамин Д. Филпот и др. «Электрофизиологический фенотип синдрома Ангельмана различается между генотипами». Биологическая психиатрия
(2019). - Jana NR (2012). «Понимание патогенеза синдрома Ангельмана через модели животных» . Нейропластичность
.
2012
: 1–10. DOI : 10.1155 / 2012/710943 . PMC 3399338 . PMID 22830052 . - Williams CA, Angelman H, Clayton-Smith J и др. (1995). «Синдром Ангельмана: консенсус по диагностическим критериям. Фонд синдрома Ангельмана». Являюсь.J. Med.Genet
.
56
(2): 237–8. DOI : 10.1002 / ajmg.1320560224 . PMID 7625452 . - Williams CA, Beaudet AL, Clayton-Smith J и др. (2006). «Синдром Ангельмана 2005: обновленный консенсус по диагностическим критериям». Являюсь.J. Med.Genet.
.
140
(5): 413–8. DOI : 10.1002 / ajmg.a.31074 . PMID 16470747 . S2CID 2449346 . - Buntinx IM, Hennekam RC, Brouwer OF и др. (Март 1995 г.). «Клинический профиль синдрома Ангельмана в разном возрасте». Американский журнал медицинской генетики
.
56
(2): 176–83. DOI : 10.1002 / ajmg.1320560213 . PMID 7625442 . - Leung, HT; Кольцо, H (январь 2013 г.). «Эпилепсия при четырех генетически обусловленных синдромах умственной отсталости». Журнал исследований интеллектуальной инвалидности: JIDR
.
57
(1): 3–20. DOI : 10.1111 / j.1365-2788.2011.01505.x . PMID 22142420 . - Andersen WH, Расмуссен RK, Стрёмма P (2001). «Уровни когнитивного и лингвистического развития при синдроме Ангельмана: исследование 20 детей». Логопедия, Фониатрия, Вокология
.
26
(1): 2–9. DOI : 10.1080 / 140154301300109044 . PMID 11432411 . - Lossie А, Дрисколл D (1999). «Передача синдрома Ангельмана от пораженной матери» . Genet Med
.
1
(6): 262–6. DOI : 10.1097 / 00125817-199909000-00004 . PMID 11258627 . - Лан Л.А., ден Бур AT, Hennekam RC, Ренье WO, Брауэра (1996). «Синдром Ангельмана в зрелом возрасте». Являюсь.J. Med.Genet
.
66
(3): 356–60. DOI : 10.1002 / (SICI) 1096-8628 (19961218) 66: 3 <356 :: AID-AJMG21> 3.0.CO; 2-K . ЛВП : 2066/22929 . PMID 9072912 . - Coppus, Antonia МВт (2013). «Люди с умственной отсталостью: что мы знаем о зрелости и продолжительности жизни?». Обзоры исследований нарушений развития
.
18
(1): 6–16. DOI : 10.1002 / ddrr.1123 . PMID 23949824 . - Steffenburg S, Gillberg CL, Steffenburg U, Kyllerman M (1996). «Аутизм при синдроме Ангельмана: популяционное исследование». Педиатр.Neurol
.
14
(2): 131–6. DOI : 10.1016 / 0887-8994 (96) 00011-2 . PMID 8703225 . - Петерсен MB, Brøndum-Nielsen K, Hansen LK, Wulff K (1995). «Клиническая, цитогенетическая и молекулярная диагностика синдрома Ангельмана: оценочный уровень распространенности в датском графстве; заболевание преимущественно поражает англосаксов». Являюсь.J. Med.Genet
.
60
(3): 261–2. DOI : 10.1002 / ajmg.1320600317 . PMID 7573182 . - Galassi FM, Armocida E, Rühli FJ (сентябрь 2016). «Синдром Ангельмана в детском портрете с рисунком Джованни Ф. Карото» (PDF) . JAMA Pediatr
.
170
(9): 831. DOI : 10,1001 / jamapediatrics.2016.0581 . PMID 27380555 . CS1 maint: использует параметр авторов ( ссылка ) - Уильямс, Чарльз. «Гарри Ангелман и история AS» . Будьте в курсе
. США: Фонд синдрома Ангельмана. Архивировано из оригинала на 2011-06-30 . Проверено 1 июля 2011 . - Дули, JM; Berg JM; Pakula Z; МакГрегор DL. (1981). «Кукольный синдром Ангельмана». Am J Dis Child
.
135
(7): 621–4. DOI : 10,1001 / archpedi.1981.02130310027010 . PMID 7246489 . - Уильямс, Калифорния; Фриас JL (1982). «Синдром Ангельмана (« счастливой марионетки »)». Am J Med Genet
.
11
(4): 453–60. DOI : 10.1002 / ajmg.1320110411 . PMID 7091188 . - Magenis, RE; Коричневый MG; Lacy DA; Budden S; ЛаФранки С. (1987). «Синдром Ангельмана является альтернативным результатом del (15) (q11q13)?». Am J Med Genet
.
28
(4): 829–38. DOI : 10.1002 / ajmg.1320280407 . PMID 3688021 . - Тан, WH; Берд, Л. М. (июнь 2021 г.). «Фармакологические методы лечения синдрома Ангельмана». Wiener medizinische Wochenschrift
.
167
(9–10): 205–218. DOI : 10.1007 / s10354-015-0408-Z . PMID 26758979 . S2CID 5959954 .
Синдром лиссэнцефалии Миллера — Дикера
При синдроме лиссэенцефалии Миллера – Дикера причиной патологических изменений является делеция некоторых генов в локусе 17p13. При этом больше всего страдает центральная нервная система. Наряду с лиссенцефалией (сглаживание находящихся на поверхности мозга извилин из-за нарушения деятельности гена PAFAH1B1) отмечается сокращение числа кортикальных слоев. Если в норме их насчитывается 6 штук, то у больных можно обнаружить только 4. Сопутствующими признаками является заметное изменение форм лица. Кроме того, больные медленно растут. Попытки интеграции в общество осложняются множественными патологиями сердца, желудочно-кишечного тракта, почек. Если при заболевании происходит делеция гена 14-3-3 эпсилон, то синдром проявляется значительно тяжелее.
Особенности воспитания
Клинические проявления недуга с возрастом могут исчезать, изменяться, сменяя друг друга. Все зависит от того, в каких условиях и эмоциональной обстановке проживает больной малыш. Дети, страдающие данным заболеванием, нуждаются в особом отношении со стороны родителей.
В частности, никогда нельзя акцентировать внимание ребенка на том, что он болен, ущербен, отличается от других детей.
Больной ребенок также, как и все остальные дети, нуждается в любви, ласке, понимании со стороны дорогих ему людей.
Конечно, чтобы привить ребенку элементарные навыки и хоть какую-то самостоятельность, родителям придется немало потрудиться, ведь действия, которые считаются обычными для других детей, у больного ребенка вызывают серьезные затруднения.
Однако, если соблюдать рекомендации лечащего врача, регулярно проводить с ребенком специальные физические и обучающие занятия, шанс на то, что малыш проживет более- менее комфортную жизнь, существенно увеличивается.
Аниридия
При аниридии нарушается нормальное строение глаза: в органе зрения отсутствует радужная оболочка. Кроме того, часто развиваются сопутствующие патологические изменения, такие как макулярная гипоплазия и гипоплазия зрительного нерва, изменения роговицы, катаракта. Острота зрения заметно падает, попытки коррекции не приносят существенных результатов. Развивается светобоязнь и горизонтальный нистагм. В некоторых случаях отмечается появление врожденной глаукомы.
Причиной заболевания является нарушение функционирования гена PAX6 из короткого плеча 11 хромосомы. Кодируемый им белок приводит к запуску ряда процессов, которые управляют процессом правильного формирования органов зрения и ряда других структур. Примечательно, что ген очень консервативен: отличие форм PAX6 у человека и данио рерио составляет менее 5%, несмотря на расхождение эволюционных линий примерно 400 млн лет назад.
Заболевание относится к группе аутосомно-доминантных патологий. В случае гомозиготности по мутантной копии гена PAX6 негативный эффект на организм возрастает, что вызывает множественные нарушения в работе органов зрения. Кроме того, поражается ЦНС, что приводит к летальному исходу.
Лечение направлено на сглаживание симптомов. Для визуальной имитации зрачка рекомендуется использовать специальным образом окрашенные линзы. Возможно восстановление зрачка путем реконструктивной пластической операции.
Клиническая характеристика ребенка
Внешне больной ребенок выглядит вполне довольным и счастливым, однако, это впечатление обманчиво.
У таких детей всегда отмечается сильная задержка умственных и физических показателей, что накладывает свой негативный отпечаток на обучение и общение со сверстниками.
В тяжелых случаях развивается стойкое и значительное нарушение моторики рук, что делает ребенка неспособным самостоятельно о себе заботиться (в частности, ребенок не может самостоятельно застегнуть пуговицу или молнию на одежде или обуви, совершать какие-либо другие действия, которые здоровому человеку кажутся вполне обыденными).
Все это существенно ухудшает качество жизни ребенка.
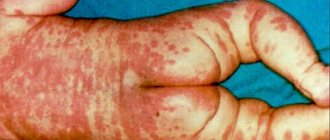
У детей, страдающих синдромом Ангельмана, отмечаются и внешние отличия, такие как:
- плоское лицо (средняя его часть), недостаточное развитие лицевых костей черепа;
- маленький и заостренный подбородок;
- гипертрофированная нижняя челюсть, которая сильно выдается вперед;
- большой и широкий язык, который ребенок часто высовывает наружу;
- нарушения зубного ряда, наличие межзубных промежутков;
- маленький череп (остальные кости скелета имеют нормальные размеры, соответствующие возрастным показателям);
- деформация (искривление) позвоночника.
Синдром Ди Джорджи
При синдроме Ди Джорджи у больных отмечается наличие врожденной формы аплазии паращитовидных желез и тимуса. Является разновидностью идиопатического изолированного гипопаратиреоза. Встречается достаточно редко.
При этом заболевании патологические изменения касаются околощитовидных (паращитовидных) желез, у которых отмечается дисгенез или агенезия. Вилочковая железа (тимус) отсутствует от рождения. В результате сочетания таких патологий происходит резкое снижение числа Т-лимфоцитов, формируется иммунологическая недостаточность. Кроме того, этот синдром сопровождается формированием врожденных аномалий крупных сосудов.
Заболевание является аутосомным и определяется наличие мутации в 22 хромосоме. В большинстве случаев причиной является спорадическая делеция 22q11 (реже микроделеция 22q11.2). Наследование происходит по доминантному принципу, с полом не связано. Некоторые авторы не соглашаются с такой характеристикой и приводят аргументы в пользу аутосомно-рецессивного типа, обладающего различной эспрессивностью.
Для заболевания характерно нарушение процесса эмбриогенеза 3-4 жаберных карманов, что приводит к нарушению закладки вилочковой железы и паращитовидных желез.
В клинике наиболее постоянными симптомами являются кандидомикоз и гипопаратиреоз, довольно часто сопровождающиеся нарушением процесса формирования рта, носа и ушей.
Тимус из-за нарушения развития в эмбриональном периоде остается неразвитым. Эпителий тимуса не обеспечивает нормального процесса развития Т-клеток. В итоге формируется специфическая форма иммунодефицита, при которой ослабляется гуморальный иммунный ответ и ответ на клеточном уровне. Если у ребенка имеется подобное патологическое нарушение иммунитета, то он будет обладать повышенной чувствительностью к инфекциям бактериального, вирусного и грибкового происхождения.
Синдром может протекать в форме генетически обусловленного отсутствия паращитовидных желез или изолированной недостаточности околощитовидных желез – в сопровождении гипокальциемических судорог, которые начинаются от рождения. Иммунологическая недостаточность приводит к появлению различных инфекционных заболеваний. Как правило, совокупность симптомов вызывает сердечную недостаточность. Кроме того, летальный исход вызывают инфекционные болезни.
Диагностика синдрома предполагает выявление типичных для синдрома патологий: искажения формы лица и черепа, наличие иммунологической недостаточности, аплазии тимуса, дисгенезии или агенезии паращитовидных желез. Ярче всего при заболевании проявляются кандидомикоз и гипопаратиреоз.
Формы
Синдром Ангельмана связан с четырьмя вариантами генетических мутаций:
- Вновь возникшей хромосомной мутацией, которая связана с потерей участка хромосомы в локусе 15 q11 — q13 . Данная мутация – причина около 80 % всех случаев заболевания.
- Одноотцовской дисомией, которая связана с потерей материнского локуса (отсутствие генетического материала матери). Данный вариант встречается редко (около 5% всех случаев).
- Дефектом ряда генов, подверженных геномному импринтингу (ГИ). Данные дефекты возникают у 2-4% больных в результате непосредственного нарушения импринтинга (различия в преобразовании информации гена в белок или РНК, которые зависят от происхождения гена). Чаще всего возникает в результате утраты центра регуляции ГИ. Дефекты ГИ без утраты центра регуляции являются результатом спонтанной мутации, повторение которой – большая редкость.
- Спонтанной мутацией материнской копии, которая вызывает отсутствие преобразования в мозге копии гена UBE3A. Данный ген кодирует деятельность убиквитинлигазы (фермент, участвующий в сложном процессе распада белков). Дефицит данного фермента относится к молекулярным механизмам синдрома.
Установить форму заболевания у 7-9 % в настоящее время не представляется возможным.
Ретинобластома
Ретинобластомой называют злокачественную опухоль сетчатки глаза. Процесс развития начинается обычно в детском возрасте, причем исходным материалом являются ткани эмбрионального происхождения. Пиковая фаза приходится на двухлетний возраст.
Практически все известные случаи выявляются в течение первых 5 лет жизни.
Причиной заболевания в большинстве случаев является мутация в генетическом материале. При этом необходимо наличие генетической обусловленности за счет наличия мутантной версии гена Rb, полученной по наследству. Вторая мутация, вызывающая появление опухоли, происходит в ретинобласте.
Существует небольшая вероятность, что у родителей, которые переболели ретинобластомой, могут родиться дети с отсутствием патологического изменения.
Отмечаются односторонние и двусторонние случаи ретинобластомы. По статистике для двусторонней формы вероятность наследственного происхождения заметно выше.
Симптомы заболевания включают боль в глазах, свечение зрачка, а также потерю зрения. Выявить их у маленького ребенка очень и очень трудно.
Диагностика обычно проходит в форме обследования под наркозом с применением УЗИ, КТ и МРТ. Достаточно распространенным приемом является биопсия красного костного мозга и спинномозговая пункция. По тяжести симптомов выделяется 5 групп.
Существует два эффективных метода лечения. При криотерапии и фотокоагуляции остается возможность сохранить и зрение, и сам глаз. Осложнения при их использовании возникают редко. Тем не менее, если возникнет рецидив, лечение потребуется повторить в той же форме. Обычно криотерапия используется в случаях, когда поврежден передний отдел сетчатки. Для заднего отдела более предпочтительным вариантом представляется фотокоагуляция.
Лечение и профилактика
Влиять на генетический дефект невозможно, нарушения происходят на хромосомном уровне, поэтому больным назначают симптоматическое лечение и психологическую коррекцию.
Медикаментозная терапия может применяться при эпилептических приступах. Для смягчения симптомов заболевания и улучшения сна могут использоваться успокаивающие препараты.
Разрабатываются индивидуальные программы для каждого пациента – ЛФК, логопедические занятия, лечебный массаж, поведенческая терапия. Они дают возможность улучшить качество жизни человека. Также привлекают логопедов, дефектологов, специалистов по невербальным способам общения и поведенческой терапии.
В профилактических целях парам, планирующим зачатие ребенка, рекомендуется консультация генетика.