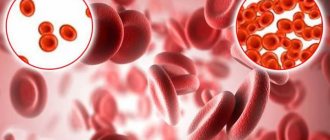
Sickle cell anemia refers to a group of genetic disorders that affect hemoglobin. The disease can be life-threatening, but there are ways to manage the symptoms. Red blood cells contain hemoglobin, which delivers oxygen to the body's tissues. In sickle cell disease, the red blood cells are C-shaped (like a sickle) and sticky. As a result, red blood cells can block blood vessels and are unable to effectively deliver oxygen, resulting in a variety of symptoms.
Genetics Home Handbook , sickle cell disease disproportionately affects “people whose ancestors come from Africa; Mediterranean countries such as Greece, Türkiye and Italy; Arabian Peninsula; India and the Spanish-speaking regions of South America, Central America and parts of the Caribbean."
Types
There are several different types of sickle cell disease. The main types include:
- HbSS
- Sickle cell disease (homozygosity for abnormal HbS): A person inherits two genes, one from each parent, for sickle cell disease. He will have sickle cell anemia, which is the most severe type of sickle cell anemia. - HbSC is a double heterozygous (compound heterozygous) carrier of two abnormal hemoglobins HbS and HbC
: a person inherits the sickle cell gene from one of the parents. A gene is inherited from the other parent that results in a different type of abnormal hemoglobin. This disease is usually less serious. - Sickle cell beta thalassemia HbS
: A person inherits the sickle cell gene from one parent and the gene for beta thalassemia, another type of anemia, from the other parent. - Carriage of the sickle cell trait
: If a person has only one gene, they will not have sickle cell disease, but they can pass the gene on to their children.
Sickle cell disease can affect anyone, but is more common in black people. Sickle cell anemia is more likely in areas where malaria is common. However, patients with sickle cell disease have a lower risk of some severe types of malaria.
Prevalence of the disease
The ethnic mixing of the population in the United States has led to a prevalence of the disease of 1 case per 5,000 population. Mutations are detected according to statistics in one baby of Negroid origin for every 500 births.
A high incidence has been established in areas that pose an epidemiological danger for malaria (Africa, India, Mediterranean countries). Up to 1/3 of the descendants of people from South Africa have altered genes. People who contain hemoglobin S in their blood do not become infected with malaria; they are not dangerous from malarial plasmodium.
Sickle cell anemia - symptoms
If the body's cells do not receive enough oxygen, various symptoms and complications develop. It can happen at any age and symptoms will vary from person to person. Sickle cells are destroyed more easily than healthy red blood cells. This can lead to low levels of red blood cells (RBCs), known as anemia.
Early symptoms may include:
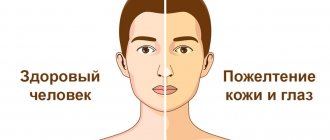
- jaundice, or yellowing of the skin and whites of the eyes
- fatigue
- pain and swelling in the arms and legs
Further symptoms and complications may include:
- episodes of pain
- swelling in the arms and legs
- acute chest syndrome
- vision loss
- enlarged spleen
- leg ulcers
- stroke
- deep vein thrombosis
- damage to the liver, heart or kidneys
- cholelithiasis (in young people)
- infertility (in men)
- pulmonary hypertension, which is high blood pressure in the blood vessels supplying the lungs
- heart failure
- damage to bones and joints that occurs due to low blood supply
- high risk of infections, which may have severe symptoms
- fever
The main symptoms and complications of sickle cell anemia:
- Pain
: During a painful episode, the sickle cells become clogged and prevent blood flow to any part of the body. The pain can range from mild to severe and can last for any period of time. - Infections
: People with sickle cell disease may be at high risk of severe illness from COVID-19, regardless of age. - Chest syndrome
- includes chest pain, cough, fever and difficulty breathing. - Enlarged spleen
: weakness, pale lips, rapid breathing and heart rate, thirst and abdominal pain are noted.
If any of the above symptoms appear, a person needs emergency medical attention.
In infants
Because a person inherits the disease, symptoms of sickle cell disease are observed before birth. A routine blood test at birth will show whether this condition is present. In severe cases, the child may develop aplastic crisis, in which the bone marrow stops producing red blood cells, resulting in severe anemia. The child may have an enlarged spleen. Symptoms include poor appetite and lethargy.
How do altered properties of hemoglobin affect hematopoiesis?
Hemoglobin S forms crystalline chains that change the shape of the red blood cell to a sickle shape, making it look like a crescent. In this form it:
- loses the ability to bind an oxygen molecule and transport it through the bloodstream;
- undergoes increased breakdown in the spleen, since the “life” of cells is shortened;
- not resistant to hemolysis (dissolves in the blood under the action of enzymes).
Impaired transportation is manifested by tissue hypoxia of internal organs. The bone marrow constantly receives “requests” for the synthesis of normal globin, so the main stem cells from which leukocytes and platelets develop are injured.
Sickle cell anemia - diagnosis
Newborn screening involves a simple blood test to detect sickle cell disease and carrier status of the sickle cell trait. If tests show that your child has sickle cell disease, doctors will monitor him over time. Tests are also available before birth, starting at 8-10 weeks of pregnancy.
If a person with a family history of sickle cell disease plans to have children, they can see a genetic counselor and undergo genetic testing to find out if their children are likely to have the disease.
Symptoms during the height of the disease
At the height of the disease, hemolytic anemia occurs constantly. In the bone marrow of tubular bones, the medulla grows, which changes the structure of the skeleton. Patients have typical changes:
- elongated (“tower”) skull and distinct bulges of the forehead;
- rachiocampsis;
- thin bones.
Compensatory activation of hemoglobin synthesis in the liver and spleen leads to their sharp increase, thrombosis of regional vessels, hemosiderosis (increased iron deposition) in the tissue of the pancreas, heart, and liver. This contributes to the development of cholelithiasis, cirrhosis, and diabetes.
In the kidneys with sickle cell anemia, small arteries of the glomeruli are thrombosed, filtration is reduced, which leads to renal failure with the accumulation of waste and nitrogenous substances.
Neurological symptoms are manifested by impaired cerebral circulation (stroke) with paralysis or paresis of muscle groups and cranial nerves.
Trophic changes in the form of non-healing ulcers on the feet and legs associated with thrombosis of the arteries of the legs are typical.
Patients endure this period for up to five years. At the same time, the mortality rate is the highest.
Sickle cell anemia - treatment
Drug treatment
The following medications may reduce the risk of complications:
- Hydroxyurea
(Hydrea): Helps provide oxygen to the body. It is not safe to use during pregnancy. Studies have not proven the benefit of this drug in children younger than 9 months. - L-Glutamine Oral Powder
(Endary): Helps reduce the number of sickle cells and is suitable from age 5. - Voxelomotor
(Oxbrita): Increases healthy hemoglobin levels. Suitable from 12 years of age. - Crizanlizumab-tmca
: Reduces pain by preventing blood cells from attaching to blood vessels. Suitable from 16 years of age.
Prevention of infections
Doctors recommend that adults and children take precautions to reduce the risk of infection. These include:
- regular hand washing
- stay away from reptiles such as turtles as they can carry salmonella
- Get vaccinations against influenza, pneumococcal disease, and meningococcal disease
Your doctor may prescribe penicillin or another antibiotic for children under 5 years of age.
Blood transfusion and stem cell transplantation
People with severe symptoms may require a blood transfusion or stem cell transplant. Blood transfusions may be necessary if the patient has severe anemia, an enlarged spleen, infection, or other complications.
A stem cell transplant using cells from a healthy donor can cure sickle cell disease, but can be risky. In addition, the stem cells must be precisely selected.
Various types of hemolytic crises
Hemolytic crisis conditions differ in the leading symptom, so it is customary to distinguish types:
- thrombotic - leading to vascular thrombosis, organ infarctions;
- hemolytic - the development of hemolysis, jaundice, a decrease in hematocrit in the blood, an increase in bilirubin prevails;
- aplastic - against the background of a threatening drop in hematocrit, the infection causes inhibition of bone marrow hematopoiesis, increased consumption of folic acid;
- sequestration - in the blood there is a general inhibition of cell growth, a sharp increase and soreness of the liver and spleen.
A rarer type is acute chest syndrome. It is characterized by a sudden increase in body temperature, chest pain, and shortness of breath. An x-ray reveals multiple infiltrates in the lungs.
The duration of crises can be up to a month.
Lifestyle
Many people with sickle cell disease can live full and active lives, especially if they take steps to reduce the effects of the condition. Lifestyle measures that may help include:
- regular medical examinations
- drink 8 to 10 glasses of water per day
- do not overcool or overheat, as this can provoke a crisis
- engage in physical activity, but also get enough rest
- maintaining a healthy diet.
Scientific article on the topic: Gene therapy will help cure sickle cell anemia.
Clinical manifestations of the initial period
Symptoms of sickle cell anemia are more pronounced when there is a high level of hemoglobin S in the blood. They manifest themselves through two mechanisms:
- increased thrombus formation in small vessels;
- manifestations of hemolysis, destruction of red blood cells.
In the initial period of the disease, the bone marrow hematopoietic system and arteries feeding the bones are affected. Symptoms appear:
- pain in the bones is first aching, then very intense, requiring the administration of analgesics;
- swelling of the legs, joints;
- increased patient anxiety;
- infection can lead to osteomyelitis.
Sickle cell anemia occurs in the form of crises, they are called hemolytic. Provoked by any acute infection. The inability to quickly restore red blood cell synthesis leads to death.
Other options:
- sequestration (storage) of blood in the liver and spleen, in the absence of hemolysis, abdominal pain appears, a sharp increase in these organs, a typical drop in blood pressure;
- Pulmonary thrombosis causes pulmonary infarction if the clotting disorder occurs at the level of the pulmonary vessels.
Mechanism of the disease.
In people with sickle cell disease, most red blood cells retain their normal disc-shaped shape. However, under certain circumstances, namely when there is a lack of oxygen, their shape changes. They become elongated, pointed and rigid, which prevents their free movement along the vascular bed and leads to blockage of blood vessels, especially the smallest ones. Due to insufficient blood supply to tissues, processes of decay and inflammation begin in them, causing pain and functional impairment. Most often, pain occurs in the limbs, abdomen, lower back or head. The most commonly affected organs include the lungs, bones, spleen, kidneys, heart and brain.
In addition to their bizarre shape, sickle cells are characterized by less strength and shorter lifespan than normal cells. Therefore, in patients with sickle cell anemia, despite the increased rate of red blood cell formation, the hemoglobin content is reduced. They appear anemic and often somewhat jaundiced (due to the rapid breakdown of red blood cells). Growth and maturation are slowed in childhood and adolescence. Patients are extremely susceptible to any infection; are less susceptible to malnutrition and dehydration because the kidneys lose their ability to retain water; surgery and childbirth involve greater risks. It should be emphasized that complications and crises arise only from time to time. Even the most severely ill patients have long periods when they feel relatively healthy and can lead a normal life.
Is it possible to prevent the disease?
There is no way to prevent chromosome damage. Spouses who know that there are cases of the disease in the family should seek genetic advice about their upcoming pregnancy. The calculated risk of producing a homozygous heir calls into question the possibility of having a child.
At the birth of a heterozygous baby it is necessary:
- provide him with proper nutrition with sufficient folic acid;
- monitor increased fluid intake;
- protect from infections.
All initial manifestations must be treated urgently. Then a milder course of sickle cell anemia is possible.
Diagnostics
When examining a patient in the initial period, you should pay attention to the paucity of manifestations with pronounced complaints of pain during crises. Diagnosis of the disease is based on blood tests. The analyzes reveal:
- low hemoglobin content, especially during hemolytic crises (less than 80 g/l);
- reticulocytosis reaches 15%;
- color index is normochromic;
- leukocytosis is high - up to 20 x 109/l;
- increased bilirubin, serum iron.
When examining red blood cells, Jolly bodies (ring-shaped nuclei) are found, and the quantitative composition of different types of hemoglobin is calculated using electrophoresis.
Under a microscope, sickle-shaped cells are visible
The sickle-shaped form of red blood cells is also revealed by a special test involving placing a drop of blood under a cover glass and completely stopping the access of air (smearing the edges with Vaseline). Such conditions simulate hypoxic conditions and cause the transformation of blood cells.
Complex genetic tests are required to determine the mode of inheritance of the disease.
Treatment and prognosis
The basic principles of therapy for sickle cell anemia are:
- sufficient pain relief;
- administration of fluid;
- decreased blood acidity (acidosis);
- administration of folic acid;
- treatment of associated infection;
- symptomatic therapy (anticonvulsants, vitamins).
Blood transfusion is not critical because it has been shown to further increase viscosity and increase thrombus formation.
There are no special drugs that affect hemoglobin synthesis yet; folic acid helps hematopoiesis
This type of anemia is currently considered an incurable disease. The average life expectancy for male patients is 42 years, for women - 48 years.
Diagnosis is important for the treatment of other concomitant diseases in this group of patients. This helps in preventing crises and prolonging the patient's life. Sometimes it is possible to prevent a severe crisis when the condition worsens by administering fluids and antibiotics on the first day.
Clinical features in patients of different ages
Doctors encounter different manifestations of sickle cell anemia in children and adults.
During the newborn period - up to three months of age, the child is no different from other children, gaining weight normally. The first symptoms are swelling and pain in the hands and feet. At 6 months, dactylitis (inflammation of individual fingers and toes) often develops. By the age of one year, weakness in the legs, curvature of the legs, and the baby’s reluctance to walk are revealed. The child is pale, the skin has a jaundiced tint.
The clinical picture is associated with thrombosis of small capillaries and impaired blood flow in the extremities. Blood clots resolve on their own, but help is needed to relieve pain. Serious complications are caused by infection. The risk of sepsis and death of the child increases sharply.
Inflammation of the digital phalanges is more common in children
After 5 years and at school age, the likelihood of sepsis is much lower. Because it has already developed its own protective immunity and antibodies to some bacteria. The problems are associated with periodic pain in the hands. In adolescence, attention is drawn to the short stature of children and delayed development of secondary sexual characteristics. Over time, they are compared with other young people.
Women with sickle cell anemia can give birth to a child, but pregnancy will require special monitoring by a hematologist and additional prevention of complications.
In adults, thrombotic manifestations are observed in the kidneys (development of renal pathology), in the retina of the eyes (visual impairment to complete blindness), in the brain (stroke), and pulmonary infarction pneumonia.
All symptoms rarely occur in one patient. Usually there is one variant of pathological changes.