March 24, 2021
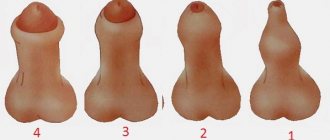
The size of the testicles is one of the indicators of the health of the genital organs, so any changes, both larger and smaller, should alert you. In normal condition, the testicles are 4-6 cm long and 2-3.5 cm wide. A decrease in size is rare, but can occur under the influence of external factors or a number of diseases. The reason why a man has small testicles can be either congenital or acquired. Let us list all possible options for the occurrence of such a condition.
Testicular hypoplasia - what is it?
The disease occurs in 5-6% of newborn boys and can be either unilateral or bilateral. The degree of hypoplasia can vary: from a slight decrease in testicular size to the complete absence of gonads. In rudimentary tissues, convoluted tubules are practically absent; underdeveloped testicles often do not even descend into the scrotum.
With unilateral testicular hypoplasia, the concentration of hormones in the body is slightly reduced, since the second organ takes over the functions of the first, increasing in size. In the bilateral form, there is a lack of testosterone and lack of fertility. Thus, with this pathology, not only is there a decrease in the organ’s size, but the formation of sperm and the production of male sex hormones are disrupted.
Varicocele
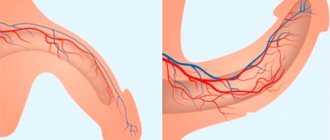
The vessels providing blood supply to the testicles have a large number of connections with each other, forming the pampiniform plexus. For some reason, the bloodstream expands. One of the common reasons why testicles become small is varicocele. This is the expansion of the veins of the spermatic cord, which provokes excess blood flow. The size reduction is about 20%.
Vasodilation occurs on the side of the veins, in 96% of cases on the left. This explains why one testicle is smaller than the other with varicocele. The size decreases with a significant disruption of the blood supply to the scrotum, when its vascular system is overfilled with venous blood. As complications, spermatogenesis may be disrupted, early male menopause or infertility may develop.
A varicocele is indicated by pain in the scrotum and discomfort when walking. Increased sweating and problems with sexual function are possible. In addition to changes in size, testicular descent on the affected side is noticeable, which leads to its asymmetry and sagging. The scrotum becomes flabby, and there is a feeling that it is “drying out.” This is because the testicle is depleted and its internal structure begins to deteriorate. With appropriate treatment, the scrotum grows to normal size.
Causes of the disease
The disease develops as a result of disturbances in embryonic development in the early stages of pregnancy. The earlier the damage occurred, the more significant the violations will be. The main cause of testicular hypoplasia is chromosomal and genetic abnormalities (damage to genes, violation of the number of sex chromosomes or their structure). An example of such a phenomenon is Klinefelter, Shereshevsky-Turner syndrome. The disease can also develop with hypogonadism, with hypoplasia of other organs of the endocrine system, adrenal tumors, alcoholic embryofetopathy, pituitary dwarfism, etc.
There are a number of unfavorable factors that can influence the likelihood of developing the disease. Pathologies of pregnancy, existing hormonally active tumors, hormonal imbalance, damage to the child’s nervous system during childbirth, etc. - these factors predispose to the development of the disease. In addition, in some cases, the cause of the pathology is an autoimmune lesion of the testicular tissue. The likelihood of the disease increases significantly if the disease is familial.
Micropenia syndrome in children and adolescents: pathogenesis, clinical picture, diagnosis
Micropenia is an abnormal decrease in penile length by more than 2.5 SD in healthy boys, which in adulthood can cause difficulty in having a full sexual life. Impaired formation of the penis in boys may be caused by structural or hormonal disorders in the hypothalamic-pituitary-gonadal system in the prenatal period. Micropenis is usually recognized soon after birth. The term "micropenis" is most often used when there is no change in the penis or scrotum.
Embryogenesis of the reproductive system
The formation of the reproductive tract in embryogenesis is determined by the interaction of three groups of factors: the genetic mechanism, internal epigenetic factors (enzyme systems, hormones) and external epigenetic factors reflecting the influence of the external environment.
The genetic sex of the unborn child is predetermined at the moment of fusion of the egg and sperm and is determined by the set of sex chromosomes formed in the zygote when the maternal and paternal gametes are combined (XX - female, XY - male), a set of special genes that primarily determine the type of gonads, and the level of activity of enzyme systems , tissue reactivity to sex hormones, concentration of sex hormones. The genetic male sex is determined by the Y chromosome (including the SRY gene, which belongs to the Sox family of DNA regulatory genes). The SRY gene encodes the regulatory factor TDF (Testis-Determining Factor). TDF causes the differentiation of the male type of gonads from the initially bipotent gonads.
Male and female gonads develop from one undifferentiated rudiment. At 3–4 weeks, the formation of the primary gonad occurs, the formation of paired Wolffian ducts, and then the Müllerian ducts. Until the 6th week of embryonic life, the embryo is morphologically the same for both female and male sexes.
At 6–7 weeks, the genital tubercle and urethral fissure, bounded by the urethral and labioscrotal folds, appear. The critical stage of development of indifferent gonads is the 8th week of intrauterine development. Under the influence of the regulatory factor TDF, as well as Sox genes, the gonadal ridges develop as testicles from the medulla of the embryo.
It has now been proven that the gene that determines the differentiation of the gonad primordium according to the male type determines the biosynthesis of a specific membrane protein, HY-antigen. The cells of the developing organism, including the cells covering the surface of the primordial gonad, contain receptors for the HY antigen. Uptake of HY antigen by these cells induces the development of the primary gonad into the testis [1].
The elements of developing testicles are spermatogonia and mesenchymal tissue. From mesenchymal cells in male embryos, Leydig cells are formed, which secrete testosterone from the 9th week under the control of gonadotropins (chorionic and pituitary). High hormonal activity of the fetal testes is necessary for the further formation of the reproductive tract in the male fetus.
The next stage of sexual formation is the differentiation of internal and external genitalia. In the early stages of embryogenesis, the reproductive system has bisexual anlages of the internal and external genitalia. The internal genital organs differentiate at 10–12 weeks of the intrauterine period. The basis of their development are the indifferent Wolffian (mesonephric) and Müllerian (paramesonephric) ducts.
During the development of a male fetus, the Müllerian ducts regress under the influence of a factor synthesized by the Sertoli cells of the fetal testicles and called “anti-Müllerian”. Wolffian ducts differentiate into epididymis, seminal vesicles, and vas deferens. The formation of the reproductive tract according to the male type is possible only in the presence of a full-fledged, active embryonic testicle.
The external genitalia are formed from the 12th to the 20th week of the intrauterine period. The basis for the development of the external genital organs of fetuses of both sexes are the genital tubercle, labioscrotal ridges and urogenital sinus.
The urogenital sinus differentiates into the prostate gland and urethra; urogenital tubercle - into the penis and cavernous bodies; genital ridges - into the scrotum. Masculinization of the external genitalia in the male fetus also consists of atrophy of the vaginal process of the urogenital sinus, fusion of the scrotal suture, enlargement of the corpora cavernosa of the penis and the formation of the male-type urethra.
The descent of the testicles from the abdominal cavity begins in the 3rd month of embryonic life, and by 8–9 months the testicles descend into the scrotum. Their descent is caused by both mechanical factors (intra-abdominal pressure, atrophy and shortening of the inguinal cord, uneven growth of the structures involved in this process) and hormonal factors (the influence of placental gonadotropins, androgens of the fetal testicles, gonadotropic hormones of the fetal pituitary gland). The descent of the testicles coincides with their maximum androgenic activity [2].
Thus, differentiation of the reproductive tract occurs during early embryogenesis, is predetermined by genetic factors and is determined by the hormonal function of the fetal gonads.
The main links in the hormonal regulation of the sexual function of the body
Conventionally, three main levels of hormonal regulation can be distinguished: a) central - cerebral cortex, subcortical formations, hypothalamic nuclei, pineal gland, adenohypophysis; b) peripheral - gonads, adrenal glands and hormones secreted by them and their metabolites; c) tissue - specific receptors in target organs with which sex hormones and their active metabolites interact.
The coordinating link of hormonal regulation is the subcortical formations and the hypothalamus, which communicate between the central nervous system, on the one hand, and the pituitary gland and gonads, on the other. A high content of biogenic amines and neuropeptides, which play the role of neurotransmitters and neuromodulators in the transformation of a nerve impulse into a humoral one, was found in the nuclei of the hypothalamus. The nuclei of the amygdala have both stimulating and inhibitory effects on the gonadotropic function of the pituitary gland, which depends on the localization of the impulse.
Of the substances found in the pineal gland, the most studied in the regulation of gonadotropic function are indole compounds - melatonin and serotonin. The inhibitory effect of melatonin on antigonadotropic function is realized at the level of the hypothalamus, blocking the synthesis and secretion of gonadotropins releasing hormone (LH-RH). In addition, other substances of a peptide nature with a pronounced antigonadotropic effect, exceeding the activity of melatonin by 60–70 times, were found in the pineal gland [3].
In the nuclei of the middle and partly posterior hypothalamus, releasing hormones are formed - substances that regulate all tropic functions of the adenohypophysis. The hypothalamus regulates sexual (gonadotropic) function through the synthesis and secretion of LH-RH gonadotropins.
The action of LH-RH stimulates the release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). The hypothalamus contains centers that carry out tonic and cyclic secretion of gonadotropins. The tonic LH-RH secretion center ensures the constant release of gonadotropic hormones. The presence of androgens is a necessary condition for the development of male-type regulation.
Three tropic hormones of the pituitary gland are directly involved in the regulation of the reproductive system: LH, FSH and prolactin (PRL). Other pituitary hormones - thyroid-stimulating hormone (TSH), somatotropic hormone (STH), adrenocorticotropic hormone (ACTH) - also affect the steroid-producing cells of the gonads.
LH is the main hormone that ensures the synthesis of the main androgen, testosterone, which is produced in the interstitial Leydig cells of the testicles. The latter, under physiological conditions, is the main inhibitor of LH secretion. Large doses of prolactin reduce the number of LH receptors [4].
Induction of steroidogenesis in Leydig cells under the influence of LH occurs when the enzyme 20a-hydroxylase is activated, which ensures the conversion of cholesterol to pregnenolone. The main androgen in the male body is testosterone. In addition to testosterone, Leydig cells produce androgens with less biological activity: dehydroepiandrosterone and Δ4-androstenedione. However, the bulk of these weak androgens are formed in the zona reticularis of the adrenal glands or serve as a product of the peripheral conversion of testosterone.
The mechanism of action of androgens on the cells of target organs is associated with the formation of the active metabolite of testosterone - dihydrotestosterone. Testosterone is converted into the active fraction directly in the cell under the influence of the enzyme 5α-reductase [2].
Hormonal regulation of the child’s reproductive system at the main stages of development
The gonadostat functions throughout the development of the child, starting from the prenatal period. In the process of developing sexual function in a child, 4 main “critical” periods can be distinguished: 1) intrauterine (fetal); 2) neonatal period; 3) prepubertal; 4) puberty.
During the fetal period, the maximum concentration of testosterone in the blood of a male fetus, characteristic of adult men, is detected between the 10th and 18th weeks of intrauterine development. This is primarily due to the participation of testosterone and its active metabolite 5α-dihydrotestosterone in the formation of the internal and external genitalia of a boy. The growth of the genital tubercle continues throughout the prenatal period.
In the first year of life, there is a sharp decrease in the level of sex hormones. Qualitative changes in the hormonal sphere occur after 6 years, when the adrenal system matures with a rapid rise in the level of adrenal androgens: dehydroepiandrosterone (DHEA) and its sulfate (DHEA-S) and Δ4-androstenedione.
The most important stage of a child’s sexual development is puberty. During this period, a complex multi-stage restructuring of the hypothalamic-pituitary relationship occurs. As the testicles increase, the size of the penis increases: first the length, and then its diameter [5].
So, the formation of external genitalia in boys during the embryonic period requires a significant amount of androgenic hormones. A decrease in testosterone biosynthesis in the embryonic testicle or a defect in 5a-reductase activity disrupts the masculinization of the external genitalia up to the formation of false male hermaphroditism.
Micropenia Clinic
The diagnosis of micropenis is made based on the correctly measured length of the penis. If the length of the elongated penis is 2.5 standard deviations less than that of the average patient with normal internal and external male genitalia, a diagnosis of micropenis is made. The normal length of the penis in a full-term boy at birth is 3.9 ± 0.8 cm, with 1.9 cm being the extreme acceptable value within 2.5 SD. By the beginning of puberty - 6.4 ± 1.1 cm, 3.7 cm, respectively; in an adult male - 13.3 ± 1.6 cm, 9.3 cm, respectively [2, 6].
The scrotum is present, although in some cases it may be underdeveloped. The testicles are located in the scrotum, but their function may be reduced. In some patients, testicular descent is impaired. Testicular volume is below normal values. Micropenia is a symptom of many congenital endocrine abnormalities occurring with hyper- or hypogonadotropic hypogonadism [7].
Klinefelter syndrome
Klinefelter syndrome (KS) is a pathology of the sex chromosome, manifested by the presence of at least one extra X chromosome (47XXY) or a mosaic karyotype. Occurs with a frequency of 1:300–1000.
Before puberty, boys with KS may exhibit cryptorchidism and a small penis. The pubertal period is characterized by gynecomastia, tall stature, and eunuchoid body proportions. The size of the testicles remains pre-pubertal, their consistency is dense. This syndrome is characterized by a 47XXY karyotype, hypergonadotropic hypogonadism (testicular hypoplasia and micropenia), infertility, reduced body hair, gynecomastia, decreased intelligence, bone deformation and high stature.
The presence of an additional X chromosome in the male karyotype does not affect the differentiation of testicles and the formation of male genitalia. However, the vital activity of germ cells is disrupted, and spermatogenesis is absent. This leads to azoospermia and infertility in adult patients [8].
XX-men
The frequency of occurrence of the XX karyotype in boys is approximately 1:20,000. The cause of the disease has not been proven in all cases. In some patients, translocation of the SRY paternal Y chromosome to the X chromosome is detected during meiosis. Phenotypically they correspond to the male sex. The penis and scrotum are formed according to the male type, but the penis can be shortened, and hypospadias occurs. The internal genital organs correspond to the male gender. The testicles at puberty remain small and dense, as in patients with Klinefelter syndrome. Adult XX men are capable of sexual activity, but they are infertile. Tallness is not typical for them, intelligence is not impaired. The levels of LH and FSH are increased.
Noonan syndrome
In 50%, a mutation in the PTPN11 gene (12q24.1) is possible, in 25% there is a mutation in the SOS gene (2p22-p21). Manifested by cryptorchidism, micropenis and scrotal hypoplasia. In addition, some patients develop a clinical picture of eunuchoidism during puberty. Often with this syndrome, pterygoid folds on the neck, a triangular face, valgus deformation of the elbow joints, short stature, lymphatic edema of the hands and feet, ptosis, sunken chest, defects of the right heart, and mental retardation are detected. Low levels of testosterone in the blood are combined with increased levels of LH and FSH.
Anorchism syndrome
Anorchism syndrome is an intrauterine, genetically determined lesion of the gonads (INSL gene mutations at 19p). At birth, the external genitalia are formed correctly, but in some cases there is a micropenis and a decrease in the scrotum. Testicles are missing. Extragenital signs of hypogonadism develop after 12–14 years. There is no puberty during puberty. Involution of secondary sexual characteristics, obesity and eunuchoidism are observed. The blood has low levels of testosterone, but high levels of LH and FSH. The test with human chorionic gonadotropin (hCG) is negative.
Gonadal dysgenesis
The cause of underdevelopment of the fetal testicle may be quantitative and structural chromosomal aberrations; a gene mutation cannot be excluded. With karyotype 46XY, structural abnormalities of the Y chromosome are possible, involving the SRY region. Dysgenetic testicles are not able to secrete active anti-Müllerian factor in sufficient quantities, and androgens do not ensure regression of the Müllerian ducts and normal masculinization of the external genitalia in the embryonic period, which causes abnormal formation of both internal and external genitalia. The external genitalia have more or less incomplete embryonic masculinization. The degree of masculinization of the urogenital sinus, as well as the size of the child’s penis, can serve as a kind of criterion in predicting the functional (androgenic) activity of dysgenetic testicles. Clinical symptoms: the patient has a dysgenetic testicle, derivatives of paramesonephric (vagina, uterus, fallopian tubes) ducts and mesonephric (epididymis) ducts. The external genitalia have a bisexual structure. Abnormal structure of the external genitalia is detected at birth
Isolated hypogonadotropic hypogonadism
With this syndrome, the secretion of LH and FSH separately or both hormones at the same time is reduced. Inherited autosomal recessively. More often caused by blocking mutations in the gonadotropin-releasing hormone receptor gene, defects in the LH or FSH b-subunit gene, respectively. The cause of impaired gonadotropin secretion is cellular insufficiency of the adenohypophysis or hypothalamus. At birth, the boy has underdevelopment of the scrotum, unilateral or bilateral cryptorchidism with inguinal dystopia of hypoplastic testicles, and micropenis. Before puberty, eunuchoidism, lack of puberty, tall stature, obesity, and gynecomastia develop. Levels of LH, FSH and testosterone in the blood are reduced [7].
A variant of the syndrome described above is congenital isolated LT deficiency (Pasqualini syndrome). Due to LH deficiency, the production of androgens in Leydig cells is disrupted. Basal and stimulated LH levels are reduced, and FSH levels are normal [3].
Kallmann syndrome
Kallmann syndrome is a genetically heterogeneous hereditary disease manifested by pituitary hypogonadism in combination with anosmia. The prevalence of the disease in boys is approximately 1:8000.
It develops as a result of disruption of the formation and migration of LH-RH neurons and olfactory nerves during the period of organogenesis, leading to anosmia and hypogonadism. Micropenis occurs with a gross defect in the secretion of LH-RH hormones. In some cases, other developmental anomalies may be present and include renal agenesis, cleft lip and/or cleft palate, selective dental agenesis, and bimanual synkinesis [9].
Prader–Willi syndrome
This is a neurogenetic multisystem disease. Develops due to damage to the critical region of chromosome 15 (segment q11.2-q13). Hypogonadism of the hypogonadotropic type may be the result of dysfunction of the hypothalamus, mainly in the region of the ventromedial and ventrolateral nuclei.
During the course of the disease, two phases can be distinguished: the first is characteristic of children 12–18 months of age. It is characterized by severe muscle hypotonia, decreased Moro, sucking and swallowing reflexes, which makes feeding the child difficult. The second comes later, after a few weeks or months. It manifests itself as polyphagia, a constant feeling of hunger, leading to the development of obesity, and fat deposition is observed mainly on the trunk and in the proximal limbs. Muscle hypotension gradually decreases and almost completely disappears by school age. The feet and hands of patients are disproportionately small - acromicria. There are also other anomalies: high, narrow forehead; almond-shaped palpebral fissures with thin, drooping eyelids; skin and hair lighter than all other family members; hypopigmentation of the iris; delayed motor development; microdontia, auricular cartilage hypoplasia, scoliosis, glaucoma. Hypogonadism (hypoplasia of the penis and scrotum, cryptorchidism) is noted. The growth of patients is often reduced [10].
Bardet–Biedl syndrome
Bardet-Biedl syndrome is a genetic pathology associated with ciliopathies. Occurs with a frequency of 1:120,000. The mode of inheritance is autosomal recessive. There are several genetic forms: BBS1 mapped to 11q; BBS2 - 16q21; BBS3 - 3p [3]. The disease is characterized by obesity, microgenitalism and cryptorchidism, oligophrenia, polydactyly, polycystic kidney disease, spastic paraplegia, and pigmentary retinopathy. LH and FSH levels are low.
Hyperprolactinemic hypogonadism
Boys in the prepubertal period develop eunuchoid body proportions: relatively long limbs, high waist, hips relatively wider than the waist of the lower limbs, fat deposits in the nipples, abdomen, and iliac crests, flabby and weak muscles, a high-pitched childish voice. The testicles and penis may be moderately hypoplastic. During puberty, secondary sexual characteristics may be absent. Sexual desire and fertility are reduced.
With hyperprolactinemia, the secretion of gonadoliberin is inhibited and the frequency and amplitude of pulse secretion of LH decreases, and the level of testosterone in the blood serum decreases. As a result of blockade of 5-alpha reductase, the conversion of testosterone to dehydrotestosterone decreases, which leads to the development of clinical signs of hypogonadism [4].
3β-hydroxysteroid dehydrogenase deficiency (3β-HSD)
This is one of the rare forms of congenital adrenal hyperplasia, which is manifested by male pseudohermaphroditism. This enzyme is involved in the biosynthesis of mineralocorticoids, glucocorticoids and sex steroids. Insufficient production of all physiologically active steroids of the adrenal cortex is associated with a defect in the HSD3B2 1p13.1 gene. This can lead to hermaphroditic external genitalia in boys. Hormonal studies reveal a decrease in the level of 17a-hydroxyprogesterone, cortisol, aldosterone, and testosterone. Before the enzyme defect, Δ5-unsaturated steroids accumulate, including large amounts of the inactive androgen DHEA. An increase in ACTH levels is typical [11].
In boys, insufficient androgenic activity of DHEA disrupts the formation of the corpora cavernosa of the penis, causing it to appear underdeveloped. During puberty, androgenization increases significantly due to the peripheral conversion of DHEA to testosterone, but does not reach the level of adult men. The latter require maintenance therapy with testosterone preparations.
17α-hydroxylase deficiency
17α-hydroxylase deficiency is caused by a defect in the CYP21 6p21/3 gene [1]. Mutations of the CYP17 gene can manifest clinically as 17a-hydroxylase deficiency, 17,20-lyase deficiency, or a combination of both. 17a-hydroxylase deficiency is characterized by a decrease in the amount, up to complete absence, of sex hormones synthesized by both the gonads and the adrenal glands, while simultaneously increasing the synthesis of mineralocorticoid precursors. In newborn boys with a defect in the CYP17 gene, the external genitalia may have a feminine structure or be slightly androgenized. At puberty, clinical symptoms of hypergonadotropic hypogonadism are characteristic. The disease is accompanied by arterial hypertension of varying severity and hypokalemia.
In 17a-hydroxylase deficiency, a decrease in cortisol stimulates the synthesis of corticotropin, and although steroid production increases, it is still blocked at the 17a-hydroxylase stage. Compensatory accumulation of 17-deoxysteroids, including pregnenolone, progesterone, deoxycorticosterone and corticosterone. Decreased androgen synthesis leads to hypogonadism. Due to high mineralocorticoid activity, hypernatremia and potassium loss occur, blood plasma volume increases, and arterial hypertension develops. Hypokalemia is usually detected, against the background of which the amount of serum aldosterone and plasma renin activity decrease. The diagnosis is confirmed by detecting a marked increase in the levels of 11-deoxycorticosterone and serum corticosterone. The amount of pregnenolone and progesterone is slightly increased. Serum concentrations of 17a-hydroxypregnenolone, 17a-hydroxyprogesterone, 11-deoxycortisol, DHEA sulfate, androstenedione and testosterone are negligible or not detectable at all. The content of FSH and LH is increased. Prenatal diagnosis is possible: determination of the content of adrenal steroids in the amniotic fluid.
17,20-desmolase (lyase) deficiency
The diagnosis of this disease is often made during puberty in individuals with a male genotype. They can be raised as girls and complain of lack of menstruation and hirsutism, or as boys and then complain of gynecomastia and underdevelopment of the genitals. In affected males, virilization rates, including clitoromegaly down to the microphallus and development of secondary male sexual characteristics at puberty, closely resemble those of 5a-reductase deficiency. All patients are infertile. The enzyme 17b-hydroxysteroid dehydrogenase or 17-ketosteroid reductase catalyzes the conversion of androstenedione to testosterone in the testicles. Mutations in the 17b-hydroxysteroid dehydrogenase type 3 isoenzyme gene (HSD17B3), located on chromosome 9q22 [2], lead to insufficient testosterone synthesis. Most of them are represented by nonsense mutations. There is no significant correlation between genotype and phenotype. Intermediate genital development is usually detected at birth. Most often there is clitoromegaly, fusion of the labioscrotal folds and a blind vaginal pouch. The testicles are often palpated in the inguinal canals or labioscrotal folds, although they can sometimes be located in the abdominal cavity. As with other forms of male pseudohermaphroditism, the internal parts of the genitourinary tract are developed normally. There are epididymis, vas deferens, seminal vesicles, and ejaculatory ducts. The prostate gland and Müllerian duct derivatives are absent. A typical laboratory finding is an elevated androstenedione/testosterone ratio, resulting from an increase in androstenedione and a decrease in testosterone. Prenatal diagnosis is possible in the offspring of affected patients if the latter have a specific mutation.
5a-reductase defect
Normally, this enzyme reduces testosterone to 5α-dihydrotestosterone (5α-DHT) in cells and, in particular, the urogenital sinus. 5α-DHT is a more active metabolite that plays a major role in the processes of differentiation of the external genitalia in boys. In its absence, normal seminal vesicles and prostate develop, but the penis and testicles are underdeveloped [11].
At birth, these boys have external genitalia similar to those of women, so most of these children are registered and raised as girls. During puberty, testosterone production increases. The architectonics of the body changes according to the male type, the total amount of bone and muscle tissue increases. The hormonal profile during this period and in adulthood corresponds to genetic sex, so many patients change their gender to male.
17-reductase defect
We are talking about an isolated disorder of testosterone synthesis. Androstenedione is not converted to testosterone.
Clinical symptoms in male patients include the manifestation of testosterone deficiency in the prenatal period (hypospadias), and later in the pubertal and postpubertal periods, symptoms of hypogonadism. However, androstenedione can be freely converted into estrogens. During puberty, gynecomastia may develop.
Testicular feminization syndrome (TFS)
A disease caused by a complete or partial lack of tissue sensitivity to androgens, due to impaired affinity of androgen receptors or post-receptor defects. Testicular feminization syndrome is one of the most common causes of false male hermaphroditism.
The disease is caused by mutations in the androgen receptor (AR) gene. Mutations cause resistance of peripheral receptors to testosterone and dehydrotestosterone. The syndrome is inherited in an X-linked recessive manner.
During embryogenesis, the gonads differentiate as fully functioning testes. However, due to a defect in the AR gene, the tissues of patients are insensitive to testosterone and dehydrotestosterone - hormones that form the male phenotype (urethra, prostate, penis and scrotum), but at the same time sensitivity to estrogen is preserved. This leads to the natural (phenomenon of autonomous feminization) formation of a female phenotype without derivatives of the Müllerian ducts (fallopian tubes, uterus and upper third of the vagina), since the production of anti-Müllerian substance by Sertoli cells is not impaired [12].
Depending on the degree of insensitivity of peripheral receptors to androgens, a complete form is distinguished (with complete insensitivity to androgens) and an incomplete form (when sensitivity is initially partially preserved or partially restored during puberty).
Morris syndrome
Clinical picture of the full form (Morris syndrome): external genitalia of the female type, with a blindly closed vagina; well-developed mammary glands (gynecomastia), absence of the uterus, fallopian tubes and prostate, absence of somatic developmental anomalies, absence of pubic and axillary hair. Gender selection is female [2].
Refeinstein syndrome
The incomplete form (Reifeinstein syndrome) is characterized by virilization of the external genitalia of varying degrees. The structure of the external genitalia reflects varying degrees of androgen deficiency during fetal development. In children with this form, high-grade hypospadias with a divided scrotum is detected, micropenis, testicles are located in the split scrotum or in the inguinal canal. Histological examination showed an increased number of Leydig cells and impaired spermatogenesis. LH and testosterone levels are elevated [2].
Hypoplasia and agenesis of Leydig cells
An autosomal recessive disease in which Leydig cell differentiation and testosterone synthesis are impaired. Occurs with a frequency of 1:1,000,000.
A point inactivating mutation in the LH receptor gene (2p21) is detected [13]. At birth, the external genitalia are formed correctly. The testicles are reduced, cryptorchidism is possible. At puberty, a clinical picture of eunuchoidism develops with obesity, underdevelopment of the external genitalia, and lack of sexual hair. The LH level is high, the concentration of testosterone and DHT is reduced, the hCG test is negative. The FSH level is normal in childhood, but decreased in puberty.
Micropenia can also develop as part of many genetic malformations not associated with sex chromosomes. Sometimes micropenis is a symptom of congenital growth hormone deficiency or congenital hypopituitarism. In some cases, the cause of micropenia cannot be determined. In such patients, endocrine dysfunction is not detected.
The size of the penis in adolescents is quite individual, so it is sometimes difficult to distinguish true micropenia associated with hypogonadism from functional one. With functional delayed sexual development, when the external genitalia have a pre-pubertal appearance, adolescents, as a rule, do not have other signs of sexual development: voice mutation, facial hair, hypertrophy of the shoulder girdle, activity of the sweat and sebaceous glands; growth retardation is observed. The basal concentration of gonadotropic and sex hormones corresponds to pre-pubertal values. Functional tests with LR-RG help in diagnosis. In response to the introduction of the latter, the release of LH increases. The bone age of such adolescents lags behind the passport age by 2–3 years.
False micropenia (hidden penis) is spoken of when the penis is of normal size, correctly formed, has well-developed corpora cavernosa and a head, but is hidden by surrounding tissues. This picture is observed in obesity - the penis is hidden in excess subcutaneous fat, but is well removed from it. During puberty, some adolescents experience hormonal dysfunction.
For the purpose of differential diagnosis, studies are carried out:
- karyotype;
- molecular genetic;
- Ultrasound of the pelvic organs;
- computer and magnetic resonance imaging of the skull;
- bone age;
- ophthalmological;
- hormonal;
- functional tests with LH-RG, with hCG [14].
For congenital micropenia, long-acting testosterone preparations are recommended, which give the best effect when administered in the first year of life. Prescribe 3-4 doses of intramuscular injections of 25 mg of testosterone cypionate or enanthate once a month. If there is no effect, the patient should be consulted by a urologist-andrologist to decide on reconstructive plastic surgery [15].
Literature
- Balabolkin M.I., Klebanova E.M., Kreminskaya V.M. Differential diagnosis and treatment of endocrine diseases (manual). M.: Medicine, 2002. 752 p.
- Dedov I. I., Semicheva T. V., Peterkova V. A. Sexual development of children: norm and pathology. M., 2002. 232 p.
- Diagnosis and treatment of endocrine diseases in children and adolescents / Ed. prof. N. P. Shabalova. MEDpress, 2009. 544 p.
- Zhurtova I. B. Hyperprolactinemia syndrome in children and adolescents: optimization of diagnostic and treatment methods. Author's abstract. dis. Doctor of Medical Sciences M., 2012.
- Guide to pediatric endocrinology / Ed. C. G. D. Brook, R.-L. S. Brown: trans. from English edited by V. A. Peterkova. M.: GEOTAR-Media, 2009. 352 p.
- Lee P.A., Mazur T., Danish R. et al. micropenis. I. Criteria, etiologies and classification // The Johns Hopkins medical journal. 1980. 146 (4): 156–163.
- Nihal Hatipoglu, Selim Kurtoglu. Micropenis: Etiology, Diagnosis and Treatment Approaches // J Clin Res Pediatr Endocrinol. 2013, Dec; 5 (4): 217–223.
- Bonomi M., Rochira V., Pasquali D., Balercia G., Jannini EA, Ferlin A. Klinefelter syndrome (KS): genetics, clinical phenotype and hypogonadism // J Endocrinol Invest. 2021, Sep 19, p. 1–12.
- Dodé C., Hardelin J.-P. Kallmann syndrome // Eur J Hum Genet. 2009, Feb; 17 (2): 139–146.
- Tozliyan E.V. Prader-Willi syndrome in pediatric practice // Pediatrician practice. 2014, March, p. 32–39.
- Dedov I. I., Peterkova V. A. Federal clinical recommendations (protocols) for the management of children with endocrine diseases. M.: Praktika, 2014. 442 p.
- Zheng Z., Armfield BA, Cohn MJ Timing of androgen receptor disruption and estrogen exposure underlies a spectrum of congenital penile anomalies // Proc Natl Acad Sci USA. 2015, Dec 29; 112(52):E7194–E7203.
- Yang-Feng TL, Seeburg PH, Francke U. Human luteinizing hormone-releasing hormone gene (LHRH) is located on the short arm of chromosome 8 (region 8 p11. 2–p21) // Somat Cell Mol Genet. 1986; 12:95–100.
- Federal clinical guidelines-protocols for the management of patients with congenital dysfunction of the adrenal cortex in childhood dated 01.2013, Moscow. 14 p.
- Nerli RB, Guntaka AK, Patne PB, Hiremath MB Penile growth in response to hormone treatment in children with micropenis // Indian J Urol. 2013, Oct-Dec; 29 (4): 288–291.
V. V. Smirnov1, Doctor of Medical Sciences, Professor A. A. Nikitin, Doctor of Medical Sciences, Professor
Federal State Budgetary Educational Institution of Russian National Research University named after. N. I. Pirogova Ministry of Health of the Russian Federation , Moscow
1 Contact information
Symptoms of testicular hypoplasia
In some patients, the disease is asymptomatic and is diagnosed only during examination for some other disease. Often the disease is detected when visiting specialists due to infertility.
Visually, the size of the scrotum is smaller than normal. The reason is a decrease in the size of one or both glands. As a rule, the size of a testicle with hypoplasia is only 0.5-2.5 cm. With unilateral damage, the difference in the size of a healthy and pathologically altered testicle can be very noticeable.
However, the severity of pathological changes is determined by the level of testosterone, i.e. The main symptom of the disease is hormonal imbalance. Often, with unilateral lesions, spermatogenesis proceeds normally; the sperm of such a man is capable of fertilizing an egg. With bilateral testicular hypoplasia, the hormonal balance is always disrupted, which leads to a lack of testosterone and, as a result, the patient develops a decrease in libido, impotence, while secondary sexual characteristics are poorly expressed. In patients over 14 years of age, axillary and pubic hair is absent or insufficiently expressed, the external genitalia are infantile, the body is disproportionate and resembles a female type, the voice is high, etc.
Hypotrophy and atrophy
A man may have small testicles due to acquired malnutrition or atrophy. It can be caused by varicocele and a number of other problems or diseases:
- testicular torsion (an acute condition that, if untreated within 6 hours, can lead to atrophy due to cessation of blood supply;
- hydrocele (testicular hydrocele, in which fluid accumulates in the scrotum);
- injuries to the groin area, surgery on the genitals or for an inguinal hernia;
- inflammatory or infectious processes (bacterial or viral orchitis, endemic mumps, sexually transmitted infections);
- testosterone replacement therapy (long-term use of hormonal drugs);
- taking anabolic drugs and estrogens;
- prolonged exposure to high or low temperatures on the scrotal area;
- hormonal imbalance, disruption of the endocrine system;
- malignant or benign testicular formations.
Surgeries on the scrotum and inguinal canal may be accompanied by compression of the spermatic artery. This leads to impaired blood supply, which provokes atrophy. Traumatic causes also include damage to the lumbar spine, causing disruption of the innervation of the testicles.
Risk factors include alcohol abuse and obesity, which contribute to the development of testosterone deficiency. The first and most important sign of atrophy is tissue death. This is the reason why a man's testicles become smaller. Their consistency changes, they become flabby, resembling a bag without contents, although sexual function is still preserved.
Due to the decrease in the size of the testicle, the amount of testosterone produced decreases, and less sperm begins to be synthesized. Atrophic processes can occur over a long period of time, and occur even at the stage of intrauterine development, which manifests itself in the form of cryptorchidism (undescended testicle into the scrotum).
Diagnostics
When the testicle is descended into the scrotum, diagnosing testicular hypoplasia is not difficult. However, this pathology is often combined with cryptorchidism - in this case, the testicle is located in the peritoneal cavity or in the inguinal canal. It can only be detected using ultrasound of the scrotum or diagnostic laparoscopy. These methods make it possible to detect not only the organ itself, but also to find out its localization, as well as to carry out a differential diagnosis with monorchism, anorchism - the complete absence of testicles, ectopia (the location of the testicle in the opposite half of the scrotum, under the skin in the groin area of the thigh, in the perineum). A biopsy is performed under ultrasound guidance.
If libido is impaired, impotence, or excess weight, testosterone levels are examined. In order to diagnose chromosomal abnormalities, a karyotype examination and genetic analysis are recommended. In adult patients, to assess the parameters of the ejaculate, it is necessary to conduct a spermogram - an analysis that allows identifying the male factor of infertility. This study must also be completed in preparation for IVF and ICSI.
How does testicular dysfunction affect procreation?
Testicular shrinkage in men is directly linked to an increased risk of infertility.
It is in the testicles that spermatogenesis occurs—the formation and development of male germ cells. Disruption of this process depends not so much on the actual reduction of the testicles, but on concomitant changes in the structure and quality of tissues of the reproductive sphere.
Clinically significant hypoplasia is almost always combined with a lack or absence of sperm, low motility and other abnormalities.
Violation of the hormonal function of the testicles also adversely affects reproductive function. Testosterone regulates many complex processes occurring both in the genitals and beyond. Even a slight imbalance in the production and utilization of sex hormones can lead to long-term infertility.
How to treat testicular hypoplasia
In most cases, treatment of testicular hypoplasia is conservative, during which stimulation and replacement therapy are performed. It should be remembered that drugs, dosage, and duration of medication are prescribed only by a doctor, and treatment tactics for each patient are selected strictly individually.
Stimulation with hormones allows you to increase sperm activity, and it is possible to achieve a therapeutic effect for quite a long time. With bilateral pathology, it is possible to develop secondary sexual characteristics with the help of replacement therapy in adolescence. In addition, with the help of hormone therapy, it is possible to significantly increase the effectiveness of IVF and ICSI. If necessary, you can use sperm cryopreservation for use in the long term.
Surgical treatment is performed for hypoplasia of one testicle, when the second one functions normally. After surgery—orchiectomy (removal)—an artificial testicle can be implanted. In some cases, an alloplastic transplant of a donor organ (transplantation) may be performed. The absolute indication for surgery is the presence of a tumor. After surgery, replacement therapy (testosterone) is prescribed.
After treatment of a one-sided form of the disease, the patient can return to sexual relations, and the possibility of conceiving a child becomes possible. With severe bilateral damage, infertility is diagnosed.
Treatment
Basically, to eliminate the problem of testicular underdevelopment, methods are used that include taking hormonal medications and hormone stimulation therapy. Depending on whether unilateral or bilateral hypoplasia is diagnosed in the patient, specific treatment is prescribed, which is accompanied by constant monitoring of the patient’s health status.
If examination and diagnosis show that one of the testicles has completely taken over the functions of the underdeveloped one, hyperplasia is treated by surgical removal of this testicle. The procedure is called orchiectomy, after which, if the patient wants it, they can perform aesthetic surgery, prosthetics or implantation of an artificial testicle.
In the event that there is an imbalance in the patient’s hormonal background with hypoplasia, a donor testicle transplantation or an alloplastic transplant is prescribed.
Prevention
Prevention of testicular hypoplasia consists of eliminating the negative effects of various negative factors on the body of a pregnant woman. In addition, special attention should be paid to the treatment of other diseases that can cause testicular hypoplasia (diseases of the endocrine system, adrenal tumors, etc.). To prevent a hereditary or genetic disease, a married couple is recommended to undergo genetic counseling. Also, in order to timely diagnose possible disorders, preventive examinations of children and adolescents by specialists such as a urologist, endocrinologist, and surgeon are necessary.
Types of services provided
| Name of service | Cost, rub. |
| Spermogram | 1 990 |
| MAR test | 1 000 |
| Testicular biopsy | 20 000 |
| Nonspecific stimulation of spermatogenesis (1 procedure) - 1st complex | 1 500 |
| Nonspecific stimulation of spermatogenesis (1 procedure) - 2nd complex | 1 900 |
| Ultrasound urological expert | 2 500 |
Subscribe to news
Simplify your life - our newsletter will be useful to you.