Porphyrias are a group of hereditary diseases that occur due to a defect in the formation of heme, resulting in the accumulation of porphyrins or their toxic precursors in the body. Heme is the iron-containing part of hemoglobin, a complex protein “responsible” for binding oxygen, transporting it to tissues and removing carbon dioxide.
During the biosynthesis of heme, compounds called porphyrins are formed. Strictly speaking, heme is a porphyrin, in the center of which there is an iron molecule Fe2+. All porphyrins are red.
Due to a defect in enzyme systems in porphyria, certain pathological metabolic products accumulate in the body. Clinical manifestations of the pathology vary depending on the specific defective gene.
Various types of porphyria occur with a frequency of 7–12 cases per 100 thousand population. Asymptomatic carriage of various genes that can lead to this pathology is approximately 50–100 people per 100 thousand1.
Classification
Heme biosynthesis occurs in the bone marrow and liver. Eight enzymes are involved in the formation of porphyrins, each of which is encoded by its own gene. Seven of them may suffer due to one or another genetic defect.
Accordingly, there are seven variants of clinical manifestations of porphyrias, which are divided into two large groups - erythropoietic and hepatic - depending on in which part of the body the predominant disruption of porphyrin synthesis is noted.
Hepatic porphyrias:
The cause of porphyrin disease is a genetic defect
- porphyria cutanea tarda
- acute intermittent porphyria
- variegated porphyria
- hereditary coproporphyria
Erythropoietic porphyrias:
- congenital erythropoietic porphyria (Gunther's disease)
- erythropoietic protoporphyria
- erythropoietic coproporphyria
Consequences and complications
Since porphyrias, with the exception of porphyria cutanea tarda, are hereditary, their cure is completely impossible. If a patient has 2-3 or more attacks per year, a neurological deficit in the form of flaccid peripheral paresis may persist for a long time. In severe cases, complications are possible in the form of bulbar syndrome , hyponatremia , paralysis of the respiratory muscles with a high risk of death. In case of porphyria with skin lesions, there is a risk of developing sclerodermoid processes.
Causes of porphyria
As already mentioned, the decrease in enzyme activity, which leads to disruption of porphyrin metabolism, is caused by a mutation in one of the seven genes encoding enzyme proteins. Moreover, even a significant (up to 50%) decrease in enzymatic activity may not cause clinical manifestations for some time. The manifestation of the disease can be provoked by:
- excessive alcohol consumption;
- some medications (non-steroidal anti-inflammatory drugs, sulfonamides, barbiturates, etc.);
- pregnancy;
- excessive insolation (sun exposure);
- starvation;
- bacterial and viral infections;
- exposure to hepatotoxic chemicals.
List of sources
- Kuznetsova N.P., Pankov B.S., Chubarova A.S. and others. Porphyria. M., 1981. p. 66–14.
- Pustovoit Y.S., Pivnik A.V., Karpova I.V. Clinic, diagnosis and treatment of porphyria // Manual for doctors. Moscow, 2003
- Vorobyov A, Kravchenko S, Kremenetskaya A, Karpova I, Pustovoit Y. Acute intermittent porphyria: problems of diagnosis and treatment // Doctor. 2003 No. 2, pp. 8-13.
- Diagnosis and treatment of acute porphyrias: Clinical recommendations of the national hematological society / ed. Pustovoit Y.S., Kravchenko S.K., Shmakova R.G., Savchenko V.G. — 2021.
- Guide to hematology in 3 volumes / ed. Vorobyova A.I. – 2005 – T.3.
Symptoms of porphyria
Clinical manifestations of porphyrias are varied and depend on the specific type of disease.
Congenital erythropoietic porphyria. The disease debuts at the age of 1–5 years. Main symptoms:
- increased sensitivity to light, up to the appearance of blisters on exposed skin;
- red color of urine;
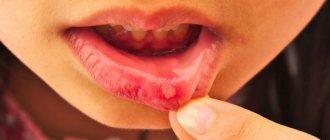
- pinkish-brown color of teeth (due to the deposition of porphyrins);
- excessive hair growth;
- enlarged spleen and hemolytic anemia (increased destruction of red blood cells).
Under the sun, blisters form on the patient's skin, which open, forming erosions, and heal with scars. Gradually the skin tightens. The mobility of the joints is impaired, the limbs may become deformed, and the phalanges of the fingers may mutate (“wither”). This is a fairly rare form of porphyria: only 100 cases are known since 1911, when the disease was first described2.
Erythropoietic protoporphyria. Manifests at 3–8 years of age. In this disease, the clinical picture is rather vague: photosensitivity manifests itself as urticaria or light pox (rashes in the form of small blisters that heal with the formation of scars). Sun rays cause itching, burning, swelling, and redness of the skin. Symptoms worsen in spring and summer and subside in winter.
Erythropoietic coproporphyria. The symptoms are similar to the previous form, but this type of porphyria is isolated because a different type of porphyrin accumulates in red blood cells.
Porphyria cutanea tarda. Most often, men aged 30–50 years old, residents of large cities with a developed chemical industry, drivers, tractor drivers, auto mechanics, and road workers suffer. The manifestation of the disease is provoked by contact with gasoline, heavy metals and other hepatotoxic compounds, as well as solar radiation and viral hepatitis. All these factors disrupt the functioning of liver enzyme systems, which provokes the manifestation of a hidden genetic defect.
Skin symptoms:
With porphyria, insolation causes the appearance of blisters and erosions
- pigmentation: the color of exposed skin varies from earthy gray to reddish-bluish;
- bullous manifestations: the reaction to the sun is similar to other types of porphyrias, insolation causes the appearance of blisters and erosions;
- hypertrichosis - increased hairiness.
Damage to the nervous system causes the following syndromes:
- asthenovegetative: weakness, sleep disturbance, increased sweating, disturbance of temperature regulation;
- polyneurotic: atrophy of the muscles of the face and shoulder girdle due to impaired innervation;
- heart rhythm disturbances: tachycardia (increased heart rate), bradycardia (decreased heart rate), arrhythmias;
- abdominal pain, constipation, diarrhea.
Hepatic manifestations:
- hepatomegaly;
- pain on palpation of the liver;
- asymptomatic sluggish hepatitis.
Damage to the eyes (conjunctivitis, retinal degeneration), ENT organs (vasomotor rhinitis) and other systems are also observed.
Acute intermittent porphyria is also characterized by a variety of clinical manifestations:
- From the gastrointestinal tract, abdominal pain (often simulating acute surgical pathologies), nausea, vomiting, constipation, diarrhea are noted.
- From the nervous system: paralysis, disorders of skin sensitivity, polyneuritis, loss of voice due to impaired innervation of the larynx, double vision (due to damage to the oculomotor nerve), urinary and fecal incontinence, epileptiform seizures
- Mental disorders: emotional lability, insomnia, depression, confusion, hallucinations
- From the cardiovascular system: hypertension, tachycardia
Mixed (variegate) porphyria. Rarely found in Europe, typical for African countries. Symptoms are similar to cutaneous tarda or acute intermittent porphyria, the only differences are biochemical.
Hereditary coproporphyria . It occurs in the form of attacks, during which abdominal pain, constipation, vomiting, depressive and hysterical manifestations are noted. Some patients experience photosensitivity. Possibly asymptomatic.
Clinical case
Patient T
., 20 years old, on December 23, 2017, during training in martial arts, he received a blow to the stomach, after which he noted an episode of nausea. After 4 days, severe pain in the epigastric region, nausea, and vomiting of bile appeared. He did not receive treatment; within 2 days the symptoms regressed. On 01/05/18, I again felt pain in the epigastric region, accompanied by vomiting and loose stools, but my body temperature did not increase. After 2 days, he was hospitalized at the district hospital at his place of residence with a diagnosis of acute pancreatitis. He denied previous illnesses and requests for medical help. Antisecretory therapy was prescribed and fasting was recommended. Over the course of 2 weeks, he lost 13 kg of body weight and developed weakness and pain in the extremities. On January 16, 2018, the patient was transferred to the intensive care unit (ICU) of the regional hospital. The examination revealed electrolyte disturbances (sodium level - 128 mmol/l, potassium - 2.8 mmol/l), amylasemia (amylase level - 223 IU/l), stenosis of the celiac trunk (80% according to computed tomographic angiography). Conservative therapy did not lead to an improvement in the patient’s condition, and therefore on January 28, 2018 he was hospitalized at the Federal Scientific and Clinical Center for Specialized Medical Care and Medical Technologies with a diagnosis of “Celiac stenosis. Chronic abdominal ischemia syndrome. Cachexia. Electrolyte disturbances" to resolve the issue of surgical treatment. Upon admission: the patient's condition is serious. The skin is of normal color and normal moisture. Body temperature 37.7 °C. Breathing is spontaneous, vesicular, with a frequency of 18 per minute, blood hemoglobin saturation with oxygen is 98%. Blood pressure - 155/110 mm Hg, heart rate - 115 beats/min. According to electrocardiography - sinus tachycardia. The abdomen was not distended, soft and painless on palpation, there were no symptoms of peritoneal irritation. He absorbed enteral nutrition, and there was a decrease in appetite and constipation for 1 week. He did not control urination; the color of the urine was straw-yellow. The patient was consulted by a vascular surgeon regarding stenosis of the celiac trunk; it was decided to refrain from surgical tactics, and conservative treatment was recommended. When examined by a neurologist: consciousness is clear, Glasgow Coma Scale (GCS) - 15 points. Orientation in place, time, and self is complete. Meningeal symptoms are negative. Pupils OD = OS, reaction to light is normal. Full movement of the eyeballs. The face is symmetrical, the tongue is in the midline. There was no dysarthria, dysphonia, or dysphagia. Muscle tone is reduced. Tetraparesis according to the muscle strength rating scale: in the arms up to 2 points in the proximal and up to 4 points in the distal sections, in the legs - up to 3 points in the proximal and distal sections. Tendon reflexes were not evoked. There were no sensory disturbances. There were pelvic function disorders such as urinary incontinence and constipation. The neurologist recommended further examination (electroneuromyography (ENMG), magnetic resonance imaging (MRI) of the brain, cervical and thoracic spine, examination of the cerebrospinal fluid) for differential diagnosis and clarification of the genesis of polyneuropathy. Laboratory examination revealed leukocytosis, mild anemia and thrombocytopenia (leukocytes 13.3 109/l, hemoglobin 12.3 g/dl, platelets 114 109/l, respectively), increased levels of pancreatic amylase (77 U/l), hypoalbuminemia (30 g/l), electrolyte disturbances (sodium 124 mmol/l, potassium 3.5 mmol/l), increased levels of inflammatory markers (C-reactive protein 47.6 mg/l, procalcitonin 7.4 ng/ml), prolongation of activated partial thromboplastin time (40 s), decrease in serum concentrations of iron (2.2 µmol/l) and folic acid (1.9 ng/ml). Computed tomography (CT) of the chest organs revealed single lesions in the lower lobes of the lungs.
Correction of water and electrolyte disturbances (infusions of sodium chloride solutions 0.9% and potassium chloride 4%) and anemia (folic acid, ferrous sulfate), empirical antimicrobial therapy (levofloxacin), mixed nutrition, prevention of stress ulcers (omeprazole), prophylaxis began thrombotic complications (enoxaparin sodium), rehabilitation measures in the ICU, including mechanotherapy in bedside simulators, physical therapy (physical therapy), placement in a bedside chair.
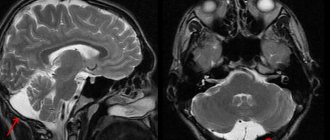
As part of the additional examination, ENMG revealed axonal demyelinating (primarily axonal) motor-sensory polyneuropathy. MRI revealed no evidence of stenosis of the cervical and thoracic spinal canals, the presence of space-occupying formations, or focal brain damage. Analysis of cerebrospinal fluid dated January 30, 2018: colorless, transparent, cytosis 2 cells/μl, glucose 3.9 mmol/l, protein 0.2 g/l; virological data (Epstein-Barr virus, cytomegalovirus, herpes simplex virus types 1 and 2) are negative. A bacteriological examination of the blood, the distal end of the removed venous catheter and sputum revealed the growth of Staphylococcus aureus, and therefore, as of January 30, 2018, levofloxacin was replaced by linezolid.
Taking into account the results of additional examination, the patient’s condition was assessed as a manifestation of Guillain-Barré syndrome and cachexia with nutritional deficiency due to pancreatitis, complicated by the development of nosocomial pneumonia, as well as catheter-associated bloodstream infection and thrombosis of the jugular and subclavian veins. On January 31, 2018, a 5-day course of specific therapy for Guillain-Barré syndrome with immunoglobulin G was started - the drug Octagam (Octapharma Pharmaceutica Production GmbH, Austria) at a dose of 0.4 mg per 1 kg of body weight per day.
Despite the treatment and daily rehabilitation measures, neurological symptoms remained at the same level. 02/05/18 pain in the limbs intensified. Considering the long history of immobilization in the ICU, the symptoms were regarded as manifestations of PTS, and fluvoxamine and carbamazepine were added to therapy. In addition, electrolyte disturbances persisted, and in order to exclude adrenal insufficiency, the patient was further examined: daily urinary sodium excretion was 87 mmol/l, serum cortisol level was 19.7 μg/dl. Taking into account the data obtained, hydrocortisone was added to therapy. Due to the ineffectiveness of linezolid monotherapy (re-growth of Staphylococcus aureus in the blood on 02/05/18, persistent high body temperature), antimicrobial therapy was intensified with cefazolin. Repeated duplex scanning revealed persistent thrombosis of the subclavian and internal jugular veins, which was probably the cause of persistent bacteremia despite antimicrobial therapy.
To exclude infective endocarditis, transesophageal echocardiography was performed, in which no evidence was obtained for the presence of vegetations on the valve apparatus and in the cavities of the heart. During esophagogastroduodenoscopy, fungal esophagitis was discovered, which was later confirmed by microscopy results.
After starting therapy with carbamazepine and fluconazole on 02/08/18, the patient’s condition showed negative dynamics in the form of a decrease in the level of consciousness to stupor (14 points on the GCS), the appearance of spontaneous nystagmus, and nausea. Repeated MRI of the brain to exclude pontine myelinolysis revealed no changes. The next day, clear consciousness was restored, but the patient became emotionally labile and began to complain of increasing aching pain in the limbs and heart, while there were no changes in the electrocardiogram.
Based on the totality of all clinical, laboratory and instrumental data, the patient was suspected of acute porphyria, which required special laboratory confirmation. On 02/09/18, a urine test revealed porphobilinogen (PBG) 200.6 mg/l, δ-aminolevulinic acid (ALA) 145.6 mg/l. The patient was transferred to the intensive care unit of the National Medical Research Center for Hematology, where he remained for 25 days. As part of the pathogenetic therapy of acute porphyria, a constant infusion of 20% glucose solution in a volume of 1000 ml/day was carried out, and a course of heme arginate (Normosang, Orphan Europe SarL, France) 125 mg/day was administered. Over time, as a result of the therapy, a decrease in the concentration of PBG from 200 to 31 mg/l and ALA from 146 to 23 mg/l was noted. The patient continued antibacterial therapy for staphylococcal infection (focal bilateral pneumonia, bloodstream infection). Pyelonephritis caused by Pseudomonas aeruginosa was diagnosed.
Treatment was carried out for thrombosis of the veins of the lower extremities and the jugular vein on the right. Due to persistent nutritional deficiency, the patient received mixed nutrition. The course of the disease in the intensive care unit was complicated by the development of symptomatic psychosis, and therefore haloperidol therapy was carried out with a positive effect. Laboratory tests revealed signs of folate deficiency and iron deficiency anemia, and folic acid replacement therapy was carried out. With the correction of hyponatremia, it was possible to stabilize the sodium level within 132 mmol/l (the minimum level reached 120 mmol/l). Kinesiotherapy and exercise therapy classes were conducted daily. On 04/04/18 the patient was discharged. His condition was regarded as moderate and was due to the course of the underlying disease, persistent flaccid paresis of the limbs, nutritional deficiency, anemia of combined origin, and thrombotic complications.
Diagnosis of porphyria
The relative rarity of the pathology and the diversity of clinical manifestations cause regular diagnostic errors. This is especially true for the diagnosis of hepatic porphyrias. Often such patients are treated by a dermatologist and other specialists to no avail.
To diagnose the disease, it is necessary to conduct an analysis to determine the level of various fractions of porphyrins in the blood, urine and feces. Depending on which porphyrin fractions the level is elevated and what the ratio between different fractions is, one or another form of porphyria is diagnosed.
Vampire disease
The connection between these two phenomena: disease and ancient beliefs about bloodsucking people was first stated by Dr. Lee Illis from Great Britain. In 1963, he presented a monograph on Porphyria and the Etiology of Werewolves to the Royal Society of Medicine. The scientist's work contained a detailed comparative analysis of surviving historical evidence that described vampires and the symptoms of porphyria. It turned out that the clinical picture of a rare disease exactly copies the portrait of the most colorful ghoul.
In advanced forms of porphyria, the skin around the lips and gums of patients dries out, causing the incisors to be exposed to the gums, creating the impression of a grin. In addition, a special substance, porphyrin, is deposited on the teeth themselves, which colors a person’s smile (or rather, grin) in a reddish-brown color. The skin on the face and body of such people becomes thinner and bursts from exposure to sunlight, becoming covered with scars and ulcers. The disease also damages cartilage, as well as the organs of which they are composed (primarily the nose and ears). Fingers become crooked. Sunlight gives the poor fellows the most severe torment, because it is under the influence of ultraviolet radiation that the breakdown of hemoglobin begins. Therefore, during the day, people suffering from porphyria try not to appear on the street, and are active only at dusk, closer to night. Either from the torment they experience, or from forced seclusion, or from some internal processes occurring in the body, these people also suffer from neuropsychic disorders and inappropriate, including aggressive behavior.
One can imagine the horror of those who one evening or night by moonlight met one of these “cute guys” on a narrow path. Here you will believe not only in vampires and werewolves, but in anything!
Treatment of porphyria
Porphyria is treated by a hematologist. Since the disease is genetic in nature, therapy is limited to reducing symptoms.
Causes and symptoms of porphyria
With erythropoietic porphyrias, sun protection comes to the fore: maximally closed clothing, wide-brimmed hats, sunscreens. To increase photoprotection, beta-carotene and nicotinic acid preparations may be recommended.
To delay the development of cirrhosis in hepatic porphyrias, patients are prescribed hepatoprotectors - for example, ursodeoxycholic acid preparations (Ursosan), which increase the resistance of hepatocytes (liver cells) to external toxic influences and thus reduce the activity of apoptosis (cell death).
To reduce the level of porphyrins in the body, bloodletting and antimalarial drugs may be recommended. Vitamins, ATP and cocarboxylase are prescribed as maintenance therapy.
About blue and simple blood
Modern scientists believe that, as often happens, innocent people suffered in the hunt for ghouls. Although the pursuers still had a reason. No, those who were accused of vampirism did not drink other people’s blood and did not turn into wild animals at night, but at the same time they looked like God forbid, and their lifestyle was, to put it mildly, suspicious. But first things first.
| But these are not fairy tales! | |
| The myth about vampires' dislike of garlic did not arise by chance. Porphyria sufferers actually cannot tolerate this spicy vegetable because the sulfonic acid released by garlic increases the damage caused by the disease in the body. But the main accusation of blood drinking is slander. It is impossible to compensate for your own red blood cell deficiency with someone else's blood. In addition, the patient’s main problem is not a lack of red blood cells in the blood, but an excess of porphyrins in the body. | |
Doctors believe that people wrongly accused of vampirism actually suffered from a rare genetic blood disorder called porphyria (from the Greek porphyros, meaning purple). It is believed that marriages between close relatives contributed to the spread of this disease. Porphyria was most common in the small villages of Transylvania (the birthplace of Count Dracula) approximately 1,000 years ago. But there are rumors that the disease has not spared the royal families either. For example, historian Andrew Wilson, in his book The Victorians, mentions hereditary porphyria, which was rampant in the British royal family, and claims that it was this disease that deprived Queen Victoria's grandfather, King George III, of his mind. However, with Victoria's accession to the throne, the crowned family got rid of this curse. Wilson believes that this was not without adultery, as a result of which the future Queen of England was born.
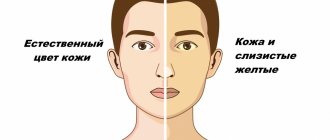
So what kind of disease is this? Today, scientists know exactly what makes people like vampires. With porphyria, the reproduction of heme, the non-protein part of hemoglobin, is disrupted, which in turn leads to excessive accumulation of toxic substances in the body - porphyrins and their precursors, which have the ability to bind metals in the body, primarily iron and magnesium. Excess porphyrins have a toxic effect on the entire body.
What is the connection between porphyria and vampires
According to medical scientists, people believed to be vampires were susceptible to a disease called porphyria, or else they had a rare genetic blood disorder. Porphyria, translated from the Greek "porphyros", means purple and is caused by consanguineous marriages, which was facilitated by low migration, especially in small villages and towns. The inhabitants of the villages of Transylvania were especially susceptible to porphyria about a thousand years ago, however, according to available information, this unusual disease did not escape the royal families.
The symptoms of porphyria have been known since time immemorial, and over time, the disease has received scientific justification for the existence of vampires - their habitats, together with their typical lifestyle and appearance, clearly indicate that the so-called vampires are simply people suffering from porphyria - about vampires and porphyria disease.
On the heredity of crowned persons
In 1998, the English publishing house Bantam Press published the book “The Purple Secret: Genes, Madness and the Royal Houses of Europe” by J. Röhl (historian), M. Warren (biochemist) and D. Hunt (JCG Röhl, MJ Warren and D. Hunt. Purple Secret: Genes, “Madness” and the Royal Houses of Europe), which explored the role of porphyria in the genealogical labyrinth of royal dynasties. King George III of England suffered from acute intermittent porphyria, but later, when indications of increased sensitivity of the king’s skin to sunlight were discovered, the diagnosis was clarified to variegated porphyria.
From the book by Mark Falkirk: “porphyria,” the symptoms of which are loss of energy, pale skin and photophobia. Then the skin becomes thinner and begins to rot, become covered with ulcers and scars, the mucous membrane of the gums and the inside of the lips bursts, begins to bleed and exposes the roots of the teeth. For many centuries, patients with this disease were mistaken for vampires and werewolves, tortured and executed.
- [1]
- Porphyria – Hematological Scientific Center of the Russian Academy of Medical Sciences
- Porphyria - history of study
- Interregional Charitable Public Organization of Disabled People “Union of Patients and Patient Organizations for Rare Diseases” - support in case of refusals to provide necessary treatment, advisory and/or legal support for patients and doctors will be provided upon receipt of a written request through the organization’s website.
- Russian Association of Porphyria. Additional information about the disease, treatment and the work of the specialized public organization of patients
- Gunther's disease
Porphyria – additional development factors
Although porphyria has a genetic basis, in only 20% of cases the genetic factor alone is a sufficient determinant of a person's clinical symptoms. In most cases, additional factors are responsible for the occurrence of the disease.
The most important ones are listed below:
- viral liver diseases,
- alcohol,
- long-term therapy with nonsteroidal drugs,
- industrial paints and varnishes,
- organic solvents,
- heavy metals,
- plant protection products,
- calorie content
- stress
- Women also have hormonal disorders.
About
Mechanisms of disease
So, vampires don't exist in real life. With this pathology, the production of the heme-non-protein part of hemoglobin is disrupted, which leads to an excess in the blood of toxic porphyrins that bind metals in the body, in particular iron and magnesium. Excessive amounts of the substance poison almost the entire body. There are several types of porphyrias, and all but the most advanced form, which affects the skin, are genetic. The modes of inheritance of each species vary, but the autosomal dominant mechanism comes to the fore.