Autoimmune hemolytic anemia
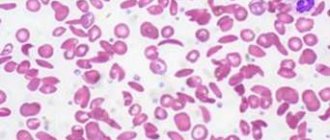
With the acute onset of autoimmune hemolytic anemia, patients experience rapidly increasing weakness, shortness of breath and palpitations, pain in the heart, sometimes in the lower back, fever and vomiting, intense jaundice. In the chronic course of the process, a relatively satisfactory state of health of patients is noted even with deep anemia, often pronounced jaundice, in most cases an enlargement of the spleen, sometimes the liver, alternating periods of exacerbation and remission. Anemia is normochromic, sometimes hyperchromic; during hemolytic crises, severe or moderate reticulocytosis is usually observed. Macrocytosis and microspherocytosis of erythrocytes are detected in the peripheral blood, and normoblasts may appear. ESR is increased in most cases. The content of leukocytes in the chronic form is normal; in the acute form, leukocytosis occurs, sometimes reaching high numbers with a significant shift in the leukocyte formula to the left. The platelet count is usually normal. In Fisher-Evens syndrome, autoimmune hemolytic anemia is combined with autoimmune thrombocytopenia. In the bone marrow, erythroposis is enhanced, and megaloblasts are rarely detected. In most patients, the osmotic resistance of erythrocytes is reduced, which is due to a significant number of microspherocytes in the peripheral blood. The bilirubin content is increased due to the free fraction, and the content of stercobilin in the feces is also increased.
Incomplete heat agglutinins are detected using a direct Coombs test with polyvalent antiglobulin serum. With a positive test, using antisera to IgG, IgM, etc., it is clarified which class of immunoglobulins the detected antibodies belong to. If there are less than 500 fixed IgG molecules on the surface of red blood cells, the Coombs test is negative. A similar phenomenon is usually observed in patients with a chronic form of autoimmune hemolytic anemia or those who have suffered acute hemolysis. Cases when antibodies belonging to IgA or IgM (against which polyvalent antiglobulin serum is less active) are fixed on red blood cells are also Coombs-negative. In approximately 50% of cases of idiopathic autoimmune hemolytic anemia, antibodies to one’s own lymphocytes are detected simultaneously with the appearance of immunoglobulins fixed on the surface of red blood cells.
Hemolytic anemia due to warm hemolysins is rare. It is characterized by hemoglobinuria with black urine, alternating periods of acute hemolytic crisis and remissions. Hemolytic crisis is accompanied by the development of anemia, reticulocytosis (in some cases thrombocytosis) and an enlarged spleen. There is an increase in the level of free bilirubin fraction and hemosiderinuria. When treating donor red blood cells with papain, it is possible to detect monophasic hemolysins in patients. Some patients have a positive Coombs test.
Hemolytic anemia caused by cold agglutinins (cold hemagglutinin disease) has a chronic course. It develops with a sharp increase in the titer of Cold hemagglutinins. There are idiopathic and symptomatic forms of the disease. The leading symptom of the disease is excessive sensitivity to cold, which manifests itself in the form of blueness and whiteness of the fingers and toes, ears, and tip of the nose. Peripheral circulatory disorders lead to the development of Raynaud's syndrome, thrombophlebitis, thrombosis and trophic changes up to acrogangrene, sometimes cold urticaria. The occurrence of vasomotor disorders is associated with the formation of large intravascular conglomerates of agglutinated erythrocytes during cooling, followed by spasm of the vascular wall. These changes are combined with increased predominantly intracellular hemolysis. In some patients, enlargement of the liver and spleen occurs. Moderately expressed normochromic or hyperchromic anemia, reticulocytosis, a normal number of leukocytes and platelets, an increase in ESR, a slight increase in the level of the free fraction of bilirubin, a high titer of complete cold agglutinins (detected by agglutination in a saline medium), and sometimes signs of hemoglobinuria are observed. Characteristic is the agglutination of erythrocytes in vitro, which occurs at room temperature and disappears when heated. If it is impossible to perform immunological tests, a provocative test with cooling takes on diagnostic significance (in the blood serum obtained from a finger tied with a tourniquet after lowering it into ice water, an increased content of free hemoglobin is determined).
With cold hemagglutinin disease, in contrast to paroxysmal cold hemoglobinuria, hemolytic crisis and vasomotor disturbances arise only from hypothermia of the body and hemoglobinuria, which began in cold conditions, ceases when the patient moves to a warm room.
The symptom complex characteristic of cold hemagglutinin disease can occur against the background of various acute infections and some forms of hemoblastosis. In idiopathic forms of the disease, complete recovery is not observed; in symptomatic forms, the prognosis depends mainly on the severity of the underlying process.
Paroxysmal cold hemoglobinuria is one of the rare forms of hemolytic anemia. It affects people of both sexes, most often children.
Patients with paroxysmal cold hemoglobinuria may experience general malaise, headache, body aches and other unpleasant sensations after exposure to cold. Following this, chills begin, the temperature rises, nausea and vomiting are noted. Urine turns black. At the same time, jaundice, enlarged spleen and vasomotor disturbances are sometimes detected. Against the background of a hemolytic crisis, patients exhibit moderate anemia, reticulocytosis, increased content of the free fraction of bilirubin, hemosiderinuria and proteinuria.
The final diagnosis of paroxysmal cold hemoglobinuria is established on the basis of the detected biphasic hemolysins using the Donath-Landsteiner method. It is not characterized by autoagglutination of erythrocytes, which is constantly observed in cold hemagglutination disease.
Hemolytic anemia caused by erythropsonins. The existence of autoopsonins to blood cells is generally accepted. In acquired idiopathic hemolytic anemia, liver cirrhosis, hypoplastic anemia with a hemolytic component and leukemia, the phenomenon of autoerythrophagocytosis was discovered.
Acquired idiopathic hemolytic anemia, accompanied by the positive phenomenon of autoerythrophagocytosis, has a chronic course. Periods of remission, sometimes lasting a considerable time, are replaced by a hemolytic crisis, characterized by icterus of the visible mucous membranes, darkening of the urine, anemia, reticulocytosis and an increase in the indirect fraction of bilirubin, sometimes an enlargement of the spleen and liver.
In idiopathic and symptomatic hemolytic anemia, the detection of autoerythrophagocytosis in the absence of data indicating the presence of other forms of autoimmune hemolytic anemia gives grounds to classify them as hemolytic anemia caused by erythropsonins. The diagnostic test of autoerythrophagocytosis is carried out in direct and indirect versions.
Immunohemolytic anemia caused by drug use. Various medicinal drugs (quinine, dopegit, sulfonamides, tetracycline, ceporin, etc.), capable of causing hemolysis, form complexes with specific heteroantibodies, then settle on erythrocytes and add complement, which leads to disruption of the erythrocyte membrane. This mechanism of drug-induced hemolytic anemia is confirmed by the detection of complement on the erythrocytes of patients in the absence of immunoglobulins on them. Anemia is characterized by an acute onset with signs of intravascular hemolysis (hemoglobinuria, reticulocytosis, increased content of the free bilirubin fraction, increased erythropoiesis). Acute renal failure sometimes develops against the background of a hemolytic crisis.
Hemolytic anemias that develop when penicillin and methyldopa are prescribed proceed somewhat differently. Administration of 15,000 or more units of penicillin per day can lead to the development of hemolytic anemia, characterized by intracellular hyperhemolysis. Along with the general clinical and laboratory signs of hemolytic syndrome, a positive direct Coombs test is also detected (the detected antibodies are classified as IgG). Penicillin, by binding to the red blood cell membrane antigen, forms a complex against which antibodies are produced in the body.
With long-term use of methyldopa, some patients develop hemolytic syndrome, which has features of the idiopathic form of autoimmune hemolytic anemia. The detected antibodies are identical to warm agglutinins and belong to IgG.
Hemolytic anemia caused by mechanical factors is associated with the destruction of red blood cells as they pass through altered vessels or through artificial valves. The vascular endothelium changes in vasculitis, malignant arterial hypertension; At the same time, platelet adhesion and aggregation are activated, as is the blood coagulation system and thrombin formation. Widespread blood stasis and thrombosis of small blood vessels (DIC syndrome) develop with traumatization of red blood cells, as a result of which they fragment; Numerous fragments of red blood cells (schistocytes) are found in the blood smear. Red blood cells are also destroyed when they pass through artificial valves (more often during multivalve correction); Hemolytic anemia has been described in the setting of a senile calcified aortic valve. The diagnosis is based on signs of anemia, an increase in the concentration of free bilirubin in the blood serum, the presence of schistocytes in a peripheral blood smear and symptoms of the underlying disease that caused mechanical hemolysis.
Less common in clinical practice is hemolytic anemia caused by exposure to lead, poisoning with acids, snake venoms or vitamin E deficiency, as well as intracellular parasites. Hemolytic anemia develops, for example, after a snake bite, accidental or intentional (suicide) ingestion of acetic acid, upon contact with lead vapor, against the background of malaria. Anemia is normocytic, normochromic, regenerative in nature; in the blood serum the content of the free fraction of bilirubin and iron is increased.
Hemolytic-uremic syndrome (Moschkovich's disease, Gasser's syndrome) can complicate the course of autoimmune hemolytic anemia. A disease of an autoimmune nature is characterized by hemolytic anemia, thrombocytopenia, and kidney damage. Disseminated damage to blood vessels and capillaries is noted, involving almost all organs and systems, and pronounced changes in the coagulogram characteristic of DIC syndrome.
Publications in the media
Hemolytic anemias are a large group of anemias characterized by a decrease in the average lifespan of red blood cells (normally 120 days). Hemolysis (destruction of red blood cells) can be extravascular (in the spleen, liver or bone marrow) and intravascular. General signs are severe intoxication with chills and fever, pain in the lower back and abdomen, possible shock as a result of impaired microcirculation, jaundice, splenomegaly, hemoglobinuria.
Etiology. Hemolytic anemia occurs due to defects in red blood cells (intracellular factors) or under the influence of causes external to the red blood cells (extracellular factors). Typically, intracellular factors are inherited, and extracellular factors are acquired.
• Extracellular factors. The microenvironment of erythrocytes is represented by plasma and vascular endothelium. The presence of toxic substances or infectious agents in the plasma causes changes in the erythrocyte wall, which leads to: (1) the production of autoantibodies to the altered erythrocyte (a classic example is autoimmune hemolytic anemia), (2) direct destruction of the erythrocyte •• Isoimmune hemolytic anemias are observed in erythroblastosis fetalis; this also includes hemolytic transfusion reactions •• Defects in the vascular endothelium (microangiopathy) can also damage red blood cells - hemolytic microangiopathic anemia. In children, it can occur acutely in the form of hemolytic-uremic syndrome •• Paroxysmal cold hemoglobinuria •• Prescription of certain drugs (for example, sulfonamides, antimalarial drugs) leads to a hemolytic crisis.
• Intracellular factors. Intracellular defects include abnormalities of red blood cell membranes, Hb, or enzymes. These defects are inherited (excluding paroxysmal nocturnal hemoglobinuria) •• Membrane defects ••• Inherited spherocytosis ••• Inherited elliptocytosis ••• Paroxysmal nocturnal hemoglobinuria •• Hemoglobinopathies (eg, sickle cell anemia). More than 300 diseases are known to be caused by point mutations in globin genes. A defect in the globin molecule contributes to the disruption of its polymerization. The membrane and shape of the red blood cell change, susceptibility to hemolysis increases •• Enzymopathies.
Some hemolytic anemias caused by enzyme deficiency
• Anemia due to deficiency of glucose-6-phosphate dehydrogenase (G-6-PD, see also appendix to this article). Clinical picture: acute episodes of hemolytic anemia, usually provoked by drugs, infections (also with neonatal jaundice and favism). Treatment • Replacement therapy (blood transfusions) • Splenectomy for G-6-PD deficiency is useless • Patients with variants of G-6-PD deficiency associated with acute hemolytic crises should avoid taking drugs that cause hemolysis. ICD-10 D55.0 Anemia due to G-6-PD deficiency.
• Anemia due to pyruvate kinase deficiency. Pyruvate kinase deficiency is the second most common (after G6PD deficiency) inherited erythrocyte enzymopathy. Pyruvate kinase catalyzes the final step of glycolysis; the consequence of its deficiency is inadequate production of adenosine triphosphoric acid (ATP), which negatively affects the functions of red blood cells, incl. at the work of Na+-K+-ATPase - defective cells lose K+. Reticulocytes (mainly in the spleen) undergo especially intensive destruction. Clinical picture • Possible manifestations of hemolysis of varying severity; in newborns - jaundice, chronic hemolysis, splenomegaly • Symptoms of pyruvate kinase deficiency are identical to those in other chronic hemolytic anemias • Due to the selective destruction of reticulocytes, their number in the peripheral blood may not reflect the severity of anemia. Treatment: splenectomy is effective. ICD-10 D55.2 Anemia due to disorders of glycolytic enzymes.
• Hemolytic anemia also develops with Norum's disease, as well as due to deficiency of the erythrocyte form of adenylate kinase (*103000, EC 2.7.4.3, AK1 gene, 9q34.1, r), adenosine triphosphatase (*102800, ATPase, EC 3.6.1.3, r, also B), d-aminolevulinate dehydratase (*125270, porphobilinogen synthetase, EC 4.2.1.24, 9q34, ALAD gene, at least 7 mutant alleles inherited by both r and B), hexokinase 1 (EC 2.7.1.1, *142600 , locus 10q22, gene HK1, r), g-glutamylcysteine synthetase (*230450, EC 6.3.2.2, r], glutathione peroxidase 1 (EC 1.11.1.9, *138320, GPX1, 3q11–q12, r), glutathione reductase ( *138300, EC 1.6.4.2, 8p21.1, GSR gene, r), glucose-6-phosphate isomerase (*172400, EC 5.3.1.9, 19q13.1, GPI gene, Â, r), 2,3-diphosphoglycerate mutases (222800, EC 5.4.2.4, 7q31–q34, BPGM gene, r), enolases (*172430, EC 4.2.1.11, ENO1 gene), coproporphyrinogen oxidase (*121300, EC 1.3.3.3, 3q12, CPO gene, В ), pyrimidine nucleotidase (*266120, EC 3.1.3.5, gene P5N, r), triosephosphate isomerase (*190450, EC 5.3.1.1, 12p13, TPI1 gene), ferrochelatase (*177000, EC 4.99.1.1, 18q21.3, FECH gene, Â, r), phosphoglycerate kinase (*311800, EC 2.7.2.3, À [Xq13, PGKA gene]; also  [arch. 19, PGKB gene]), 6-phosphogluconolactonase (*172150, EC 3.1.1.31), phosphofructokinase (*171860, EC 2.7.1.11, 21q22.3, PFKL gene). Treatment is individualized depending on the type of hemolytic anemia (for example, splenectomy or iron replacement therapy).
ICD-10 • D55 Anemia due to enzyme disorders • D56 Thalassemia • D57 Sickle cell disorders • D58 Other hereditary hemolytic anemias • D59 Acquired hemolytic anemia
APPLICATIONS
Glucose-6-phosphate dehydrogenase (G-6-PD, EC 1.1.1.49, *305900, Xq28) is important for maintaining the intracellular content of reduced nucleotides; inherited enzyme deficiency (A, extreme polymorphism, more than three hundred allelic variants) leads to the development of anemia, favism, and chronic granulomatous disease. There are variants of the enzyme: variant A and Mediterranean • Option A is found in African Americans (rapidly degrading isoenzyme, half-life - 13 days) • Mediterranean variant is found mainly in Greeks and Italians (extreme instability of the enzyme, half-life - several hours), in fact, the activity of the isoenzyme absent • In variant A or the Mediterranean variant of G-6-PD deficiency, taking drugs that exhibit oxidative properties (sulfonamides, salicylates, phenacetin) causes an acute hemolytic crisis after 1–3 days of the incubation period. The subsequent course of the disease is different for the two variants of the enzyme •• Option A. Hemolysis of the pre-existing erythrocyte population occurs. The hemolytic crisis resolves when new populations of red blood cells with high G-6-PD activity are formed in the bone marrow •• Mediterranean variant. Hemolysis leads to the destruction of almost all red blood cells. Transfusion is indicated until the drug is completely eliminated from the body.
Favism (primaquine anemia) is an acute condition that occurs when eating certain types of legumes (for example, fava beans Vicia fava), as a result of inhalation of pollen from their flowers, as well as after taking certain drugs (primaquine, sulfonamides, salicylates, nitrofurans, vitamin derivatives K, etc.). It is found everywhere, but more often in areas where malaria is endemic. Possible in some people with hereditary G6PD deficiency (305900, G6PD gene, Xq28). Clinical picture: high body temperature, headache, abdominal pain, hemolytic anemia, eosinophilia, jaundice, diarrhea, vomiting, loss of strength and coma. Treatment is supportive • blood transfusions • folic acid • maintaining adequate diuresis • urine alkalinization. ICD-10. D55.0 Anemia due to deficiency of glucose-6-phosphate dehydrogenase. Favism.
Paroxysmal nocturnal hemoglobinuria is a chronic disease with episodes of hemolytic anemia and hemoglobinuria (mainly at night). Characteristic features include yellowness of the skin or its bronze coloration, moderate enlargement of the spleen, and sometimes the liver, macrocytosis, and anisocytosis. Rarely observed (2/1 million population). Predisposition to developing the disease (*311770, Xq22.1, PIGA gene, À). Diagnosis: episodes of morning black urine; when table sugar is added to the patient's blood, hemolysis occurs. Treatment. GCs (eg, prednisolone 20–40 mg/day) cause stable remission in 50% of patients. Plasma transfusions are not recommended, but washed red blood cells can be transfused during crises. Heparin should be used with caution, because increased hemolysis is possible. Iron supplements orally. Synonyms: Marchiafava–Miceli syndrome, Marchiafava–Miceli disease. ICD-10. D59.5 Paroxysmal nocturnal hemoglobinuria. Cold is a rare disease characterized by the occurrence of hemolysis after cold exposure (atmospheric air, cold water). Usually occurs after a viral infection. Hemolysis is induced by Donath–Landsteiner hemolysins fixed on erythrocytes. The course can be acute, progressive or recurrent. Synonyms: Donath-Landsteiner hemolytic anemia, Dressler syndrome, Harley syndrome. ICD-10. D59.6 Hemoglobinuria due to hemolysis caused by other external causes.
Stomatocytosis • I (#185020, 9q34.1, EPB72 gene [133090], Â), mutation of the stomatin gene (internal protein of erythrocyte membranes). Clinically: hemolytic anemia, stomatocytosis. Laboratory: shortening of the circulation time of erythrocytes, their increased osmotic resistance, increase in the intracellular content of sodium ions • Stomatocytosis II (*185010, Â). Clinically: hemolytic anemia, stomatocytosis, cholelithiasis, periodic jaundice. Laboratory: decreased osmotic resistance of erythrocytes, increased intracellular sodium • Cold-sensitive stomatocytosis (185020, Â). Clinically: hemolytic anemia, stomatocytosis. Laboratory: Increased autohemolysis and increased osmotic resistance of erythrocytes at 5 °C, cold hemolysis prevented by a decrease in pH or an increase in ATP content.
Aldolase deficiency. Fructose-1,6-diphosphate aldolase (triosephosphate lyase, EC 4.1.2.13, 3 isoenzymes: aldolases 1, 2 and 3, or A, B and C) is a glycolytic enzyme that catalyzes the reversible conversion of fructose-1,6-bisphosphate to glyceraldehyde 3 -phosphate and dihydroxyacetone phosphate. Aldolase A is expressed in fetal tissues and skeletal muscles (5% of total muscle protein), aldolase B in the liver, kidneys, and intestines, and aldolase A and C in nervous tissue. Hereditary diseases are known that develop as a result of deficiency of various aldolases. Aldolase A (103850, 16q22–q24, ALDOA gene, r). With enzyme deficiency, congenital non-spherocytic hemolytic anemia develops (jaundice, splenomegaly, cholelithiasis, possible short stature, delayed mental development and puberty). Aldolase B (229600, 9q22, ALDOB gene, r). When the gene is mutated, congenital fructose intolerance (fructosemia) develops. Clinically: sluggish sucking, delayed growth and development, hypoglycemia, metabolic acidosis, vomiting, hepatomegaly, liver cirrhosis, gastrointestinal bleeding, seizures, proximal tubular acidosis. Laboratory: fructosemia, hyperbilirubinemia, hyperuricemia, glucosuria, hypophosphatemia and phosphaturia, high urine pH values. ICD-10. D55.2 Anemia due to disorders of glycolytic enzymes.