Ataxia - what is it?
Features of the development of ataxia
Any movement is carried out as a result of a series of successive contractions of different muscle groups (antagonists and synergists). For alternation to be normal, the work of three elements of movement coordination is necessary:
- structures and connections of the cerebellum (the central organ of movement coordination);
- receptors that perceive the state of stretching of tendons, muscles, joint capsule and their conductors at each specific moment in time,
- impulses coming from the organ that assesses the position of the body in space (vestibular apparatus).
Depending on which of these coordination elements is affected by disturbances, a different type of ataxia develops. According to the classification, there are:
- Sensitive ataxia – disturbances in the conductors of deep muscle sensitivity;
- vestibular ataxia – damage to the vestibular apparatus;
- Cerebellar ataxia (cerebellar) – damage to the cerebellum;
- Frontal ataxia (cortical) – damage to the cortex of the frontal or temporo-occipital region.
It is customary to classify hereditary ataxias as a separate group:
- Friedreich's familial ataxia,
- cerebellar ataxia of Pierre-Marie,
- ataxia telangiectasia, or Louis-Bar syndrome.
Symptoms of congenital cerebellar ataxias
Common symptoms for all types of ataxia are ataxic manifestations . This is a violation of the coordinated work of all muscles to achieve the goal of the act of movement. Symptoms of congenital cerebellar ataxia include dysmetria —disproportionality of the effort exerted to perform a goal-directed movement. Dyssynergia is observed when the coordination of individual muscles is impaired, intentional trembling, rhythmic deviation from the correct trajectory of targeted movement, which increases as the target approaches.
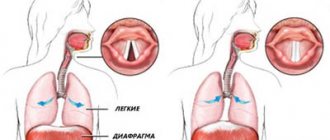
Frequent signs are instability in a vertical position, nystagmus (rhythmic rapid movements of the eyeball), as well as abrupt speech, stress on each syllable. A symptom of cerebellar ataxia in children is that the child begins to sit and walk late, and his gait is uncertain, the child seems to “sway.”
Symptoms of congenital cerebellar ataxia are manifested by a delay in the child’s motor functions, he begins to sit and walk late, there is a lag in mental development, and speech delay. Usually by the age of 10, compensation of brain functions occurs.
To confirm the diagnosis, it is necessary to conduct CT and MRI studies , as well as DNA studies to determine the genes that lead to the development of certain forms of pathologies.
Causes of ataxia
Ataxia may be a consequence of:
- severe head injury (traumatic brain injury);
- accumulation of fluid in the cavities of the brain, spinal canal (hydrocephalus);
- congenital malformation of the brain/skull;
- infectious disease of the brain (encephalitis);
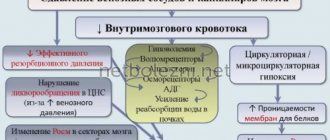
- cerebrovascular accidents (ischemic stroke);
- cerebral palsy;
- epilepsy in children;
- malignant neoplasms;
- abscesses.
Sensitive ataxia syndrome develops as a result of damage to:
- posterior brain stems,
- peripheral nodes,
- posterior nerves, optic thalamus,
- parietal lobe of the brain.
Cerebellar ataxia is a consequence of damage to the cerebellar vermis, its peduncles and hemispheres. Occurs in encephalitis, sclerosis.
The vestibular form of the disease is caused by damage to any part of the vestibular apparatus - nuclei in the brain stem, cortical center in the temporal lobe of the brain, vestibular nerve, labyrinth.
Cortical ataxia occurs when the frontal lobe of the brain is damaged, which is caused by disturbances in the functioning of the fronto-pontine-cerebellar system.
Hereditary types of ataxia are transmitted in an autosomal dominant or autosomal recessive manner.
Drug therapy
If the pathology was caused by an infectious-inflammatory process, then the doctor prescribes antiviral or antibacterial therapy. Vascular disorders that cause cerebellar ataxia are treated by stopping cerebral bleeding or normalizing blood circulation. For this, the patient is prescribed the following groups of drugs: thrombolytics, angioprotectors, antiplatelet agents, anticoagulants, vasodilators.
If cerebellar ataxia has a hereditary etiology, it is impossible to completely get rid of it. Basically, doctors resort to metabolic therapy, which involves prescribing drugs such as cerebrolysin, vitamins B12, B6 and B1, ATP, ginkgo biloba preparations, mildronate, piracetam. To increase the tone of skeletal muscles and improve metabolism in them, massage is recommended for patients.
Symptoms of ataxia
Each type of ataxia has its own symptoms. The sensitive form is characterized by a disorder of joint-muscular sensation in the lower extremities. Wherein:
- The patient is unsteady, bends his legs too much while walking, and takes loud steps.
- He has the feeling of walking on cotton wool/soft carpet.
- To compensate for impaired motor function, he tries to look at his feet.
- When closing the eyes, the symptoms become more pronounced.
- With severe damage to the posterior columns, movement becomes impossible.
Cerebellar ataxia is described by the following symptoms:
- While walking, the patient falls towards the affected cerebellar hemisphere.
- Staggers when walking, puts his feet very wide.
- The movements are awkward and sweeping.
- The severity of coordination disorder does not depend on vision control (symptoms are the same with open and closed eyes).
- Speech becomes slow, drawn out, handwriting becomes uneven and sweeping.
- Muscle tone decreases and tendon reflexes are impaired.
Vestibular ataxia is characterized by:
- dizziness, which intensifies even with smooth turns of the head,
- vomiting, nausea.
Frontal (cortical) ataxia manifests itself:
- unsteady walking (especially on turns),
- disturbances of smell,
- mental changes,
- lack of a grasping reflex.
Symptoms of hereditary ataxias
Pierre-Marie ataxia manifests itself similarly to cerebellar ataxia. It usually begins at the age of thirty-five. Then the first gait disturbances appear. Then problems arise with facial expressions, speech, and hand movements.
Tendon reflexes increase. Strength in the leg muscles decreases. Involuntary muscle twitches occur periodically. Vision deteriorates, intelligence decreases, and depressive disorders occur.
Friedreich's familial ataxia is manifested by increasing degeneration of the posterior and lateral trunks of the spinal systems, damage to the Gaulle's fascicles, Clark's columns and the posterior spinocerebellar tract. The patient develops an awkward, uncertain gait. He walks in a sweeping manner, deviates from the center to the sides, and places his feet wide.
Over time, changes affect facial expressions and speech. Periosteal and tendon reflexes decrease or disappear completely. Hearing gets worse. A severe course of the disease provokes damage to the heart, changes in the skeleton (frequent dislocations of joints, curvature of the legs, etc.).
Ataxia telangiectasia develops at an early age. It progresses quickly - by the age of ten the child can no longer walk. Accompanied by damage to the cranial nerves and mental retardation. Negatively affects the functioning of the immune system - the patient often suffers from bronchitis, pneumonia, rhinitis, sinusitis.
If you notice similar symptoms, consult a doctor immediately. It is easier to prevent a disease than to deal with the consequences.
Ataxia
Authors: Tetsuo Ashizawa and Guangbin Xia
Per. from English N.D. Firsova (2018)
Introduction
Ataxia, defined as a lack of coordination of voluntary muscle movements, is a physical symptom and not an independent disease, so its etiology should always be investigated. Ataxia may be the patient's chief complaint or one of the symptoms.
Ataxia is usually caused by cerebellar dysfunction or damage to vestibular or proprioceptive afferent input to the cerebellum. Ataxia may have an insidious onset with a chronic and slowly progressive clinical course (eg, spinocerebellar ataxias [SCA] of genetic origin) or an acute onset, especially those ataxias resulting from cerebellar infarction, hemorrhage, or infection, which can progress rapidly with catastrophic consequences. Ataxia may also have a subacute onset due to infectious or immunologic disorders, which may have a limited window of therapeutic options.
An operative strategy to address treatable causes of ataxia can save the patient's life and lead to good long-term outcomes. Ataxia may also be benign in symptomatic disorders (eg, vestibular neuritis). With the development of neurogenetics, more cases of cerebellar ataxia caused by hereditary causes are being diagnosed, but many sporadic ataxias, including those with a chronic and progressive course, still remain undiagnosed.
Symptoms and signs of ataxia
Symptoms and signs are related to the location of the lesions in the cerebellum.
Lateralized cerebellar lesions cause ipsilateral symptoms, whereas diffuse cerebellar lesions cause more generalized symmetrical symptoms. Lesions in the cerebellar hemisphere cause limb ataxia (appendicular). Worm lesions cause ataxia of the trunk and gait, with relative sparing of the extremities. Vestibulocerebellar lesions cause imbalance, dizziness, and gait ataxia (called cerebellar gait).
Acute cerebellar pathology may initially cause severe abnormalities; Over time, it may recover significantly and the disease becomes asymptomatic, even if imaging shows persistent dramatic structural changes in the cerebellum.
Chronic progressive ataxia may be associated not only with neurodegenerative or hereditary diseases of the cerebellum, but also with neoplasms and chronic infections.
Terms describing ataxia
The following clinical terms are often used to describe ataxia.
Sustainability. A healthy person can stand naturally, with their feet less than 12 cm apart, and can stand steadily with their feet together for more than 30 seconds. Poor posture in the absence of motor weakness or gross involuntary movements suggests cerebellar or sensory ataxia.
Ataxic gait . Gait ataxia results from incoordination of the lower extremities due to cerebellar pathology or loss of proprioceptive input. Patients often feel insecure and are forced to hold on to a wall or furniture and walk with their legs apart. Increasing gait disturbance when visual cues are removed (walking with eyes closed or in the dark) indicates a sensory or vestibular component of ataxia. Ataxia due to cerebellar causes remains unchanged regardless of visual findings.
Sensory ataxia . Sensory ataxia mainly manifests as gait disturbance, as described above. Additionally, subjects with sensory ataxia will have a positive Romberg's sign. They may walk with a high gait (due to motor weakness) or shuffle (to assist themselves with sensory feedback caused by sound). Pseudoathetosis (random finger movements seen on outstretched arms with eyes closed) can also occur with sensory neuronopathy affecting the upper extremities.
Trunk ataxia . Trunk ataxia may result from lesions in the midline of the cerebellum. Patients may exhibit instability in the trunk, such as body swaying while sitting (worse with arms extended forward) or standing (titubation).
Limb ataxia . The term "limb ataxia" is often used to describe upper limb ataxia resulting from incoordination and tremors. It may be better described by functional impairments such as clumsiness when writing, buttoning clothes, or trying to grasp small objects. The patient must slow down to be precise in reaching objects. Limb ataxia may be lateralized with ipsilateral cerebellar lesions.
Dysdiadochokinesis / dysrhythmokinesis . Dysdiadochokinesis, or dysrhythmokinesis, is tested by rapidly alternating hand movements or tapping the crease of the thumb with the index finger. Disorders may manifest as uneven rhythm and amplitude.
Intention tremor . Intention tremor occurs as a result of instability of the proximal limb and is manifested in an increase in the amplitude of oscillations at the end of a voluntary movement. Tested by touching your finger to your nose and your heel to your shin. This is different from essential tremor, which primarily occurs in the distal limb.
Dysmetria . Dysmetry is when the patient misses the target object due to excess (hypermetry) or insufficient (hypometry) covering the required distance. Dysmetria is often tested using the finger chase test and can be quantified by the missed distance (in cm). Dysmetria also occurs when the eyes turn off objects (ocular dysmetria); either the eyes need a second movement to catch the object (hypometric saccades) or the eyes need to correct the overshoot to focus on the object (hypermetric saccades). The shin tap test (precisely tapping the middle of the shin or knee with the heel of the opposite leg) also detects the presence of dysmetria.
Dysarthria . Dysarthria is often described by the patient or family as slurred speech. The patient's speech is irregular and slow with unnecessary fluctuations. Words are often broken into individual syllables, and some syllables with a plosive consonant are unusually stressed (chanted speech).
Nystagmus . Nystagmus often occurs in cerebellar diseases. Lateral nystagmus caused by lateral gaze is seen as a slow drift toward the midline followed by a fast saccade phase at an eccentric position. Upgaze nystagmus and downgaze nystagmus are defined by a fast phase in the up or down direction. Nystagmus when looking up is observed when the anterior part of the worm is affected. Downgaze nystagmus typically occurs when there is damage to the foramen magnum, such as an Arnold-Chiari malformation.
Saccades . The speed of saccades is usually normal in cerebellar disease, but looking too far or too close (ocular dysmetria) is often observed and is often followed by a corrective saccade in the appropriate direction. However, in SCA type 2 (SCA2) and in advanced stages of other SCAs, saccades slow down.
Square wave jerks/ocular flutter/opsoclonus . Square wave jerks, ocular flutter, and opsoclonus are terms used to describe other ocular abnormalities in cerebellar lesions. Square-wave shocks appear as two saccades in opposite directions, separated by a short period of no movement. A healthy person may have square wave motion of 0.1 to 0.3 degrees, but less than 10 per minute. Square-wave impulses with large amplitude are more characteristic of cerebellar ataxia. Ocular flutter differs from square wave jerks in that the repeated saccades are not separated by short periods of no movement. Opsoclonus is continuous conjugate saccades in all directions of a chaotic nature. Both ocular flutter and opsoclonus usually indicate cerebellar involvement in a paraneoplastic (neuroblastoma) or post-infectious syndrome (as seen in opsoclonus-myoclonus ataxia).
Neuroanatomy of ataxia
The cerebellum, its afferent and efferent connections, the vestibular system, and the proprioceptive sensory pathway are all implicated in ataxia. The cerebellum consists of the median part and the hemispheres. Lesions in each of these areas can result in a different form of ataxia. For example, damage to the midline cerebellar structures is usually accompanied by gait and trunk ataxia, whereas unilateral damage to the cerebellar hemisphere usually causes ipsilateral cerebellar ataxia. Understanding this neuroanatomy and relationship to coordination can help with localization of the injury.
Classification and etiology of ataxia
There are different ways to classify ataxia: by age of onset, rate of onset and clinical course; by anatomical involvement; focal lesion or generalized; acquired or inherited. Cases where all specific diagnoses have been excluded are classified as sporadic adult ataxia of unknown etiology, which still remains a diagnostic challenge.
Hereditary ataxia
Hereditary ataxias are rare diseases, but as diagnostic technologies develop, they are increasingly diagnosed at an earlier age. Hereditary ataxias are classified as autosomal dominant, autosomal recessive, X-linked, or mitochondrial. Characteristic features can help in recognizing a specific diagnosis.
Hereditary ataxias, especially autosomal dominant cerebellar ataxias, should be considered when the disease is transmitted vertically from one generation to another in a family. Documentation of father-to-son transmission establishes autosomal dominant inheritance.
Spinocerebellar ataxia
A family history consistent with autosomal dominant inheritance requires DNA testing for SCA, which in genetic terminology refers to a group of autosomal dominant disorders with a known chromosomal locus. Primary mitochondrial and X-linked mutations may also be considered depending on clinical presentation and family history. In the absence of a family history and secondary ataxias have been ruled out, sporadic cases may still require DNA testing, since a negative family history does not rule out hereditary disorders, and up to 5% of patients with apparently sporadic degenerative ataxia may have a positive DNA test. Due to the cost of DNA testing, health insurance must be taken into account. Although SCA has no cure, a positive DNA test result provides important practical information to patients and their families. First, it allows patients to plan for their future, such as making educational or career decisions, especially in the area of predictive testing. Second, it provides a basis for genetic counseling for patients and their families. Finally, it allows patients to participate in support group and research activities specific to the genetically defined disease. For example, genotype determined by DNA testing will be included in the criteria of most, if not all, future clinical trials of disease-modifying therapies, and once a therapy is developed and approved, patients will need to know the genotype of their disease to determine whether drug for them.
Sporadic and idiopathic ataxia in adults
For older patients with sporadic degenerative ataxia, neurologists should consider a diagnosis of multiple system atrophy of the cerebellar type (MSA-C). Documentation of involvement of other areas of the nervous system, especially the basal ganglia and autonomic dysfunction, is critical to the diagnosis of MSA-C in patients with sporadic ataxia. Brain MRI may provide clues to the diagnosis of MSA with brainstem atrophy. The remaining late-onset degenerative ataxias are often called sporadic adult ataxias, in a category known as late-onset idiopathic cerebellar ataxia. Although the two terms are used interchangeably, some experts include MSA-C in the category of late-onset idiopathic cerebellar ataxia.
Friedreich's ataxia
Friedreich's ataxia is one of the most common genetic autosomal recessive ataxic syndromes. Onset usually occurs in childhood and young adulthood, with progressive ataxia leading to loss of ambulation after 10–15 years. Other clinical features include sensory loss due to dorsal root ganglion and dorsal column degeneration with areflexia and foot deformities, scoliosis, hypertrophic cardiomyopathy, and glucose intolerance. With genetic diagnosis, the phenotype has expanded to include a later presentation (over 25 years of age and up to 60 years of age) and a slower progression called late-onset Friedreich's ataxia. These individuals have preserved reflexes, often called Friedreich's ataxia with preserved reflexes, which may show spasticity without cardiac or skeletal signs. The disease is caused by an abnormal expansion of GAA repeats in the frataxin (FXN) gene, although in rare cases point mutations may be present on one of the chromosomes instead of the GAA expansion. Age of onset is inversely correlated with the number of GAA repeats on the minor allele. Loss of frataxin function in mitochondria leads to iron-sulfur cluster deficiency, impaired oxidation, and iron accumulation. Nicotinamide has been shown to increase frataxin levels, but clinical benefit has not yet been demonstrated, and studies with the chelator deferiprone and the antioxidant idebenone are unclear regarding long-term benefit. Clinical trials have been completed with carbamylated erythropoietin, interferon gamma-1b, pioglitazone, epoetin alfa and several other substances. These studies did not provide conclusive evidence of effectiveness.
Diagnosis of ataxia
When diagnosing ataxia, the following methods are used:
- MRI of the brain (detects atrophy of the brain stems and spinal cord, the upper parts of the vermis);
- electroencephalography of the brain (diagnoses reduction of alpha rhythm, diffuse theta and delta activity);
- electromyography (reveals axonal demyelinating damage to sensory fibers of peripheral nerves);
- DNA diagnostics (used to determine hereditary types of ataxia). By conducting indirect DNA diagnostics, doctors determine whether the ataxia pathogen can be inherited by other children in the family;
- magnetic resonance angiography (allows you to assess the integrity and patency in the cranial cavity and identify brain tumors).
Additional diagnostic procedures include consultation with a neurologist, ophthalmologist, and psychiatrist. Laboratory diagnostics for ataxia show a violation of amino acid metabolism - a reduced concentration of alanine and leucine, a decrease in their excretion in the urine.
Treatment of ataxia
Independent treatment of ataxia, regardless of its type and severity of symptoms, is impossible. Consultation with a neurologist is required
.
Treatment is usually aimed at eliminating the causative disease, for example:
- tumor removal,
- elimination of hemorrhage,
- decreased pressure in the posterior fossa (Arnold-Chiari malformation),
- abscess removal,
- normalization of blood pressure.
In addition, it includes:
- selection of a special set of gymnastic exercises that help reduce incoordination and strengthen muscles;
- prescription of general strengthening agents - B vitamins, cerebrolysin, anticholinesterase drugs, ATP.
For ataxia telangiectasia, medications that correct immunodeficiency are prescribed, and a course of immunoglobulin is administered. Treatment of Friedreich's ataxia involves the use of medications that support mitochondrial function (vitamin E, riboflavin, succinic acid, coenzyme Q10).
Danger of ataxia
Ataxia is a disease in which coordination of movements is impaired. Its progression can lead to disability and even death. Tremor of the arms and legs, severe dizziness, inability to move independently, disturbances in swallowing and defecation - all these are symptoms of pathology. In addition, ataxia leads to respiratory failure, chronic heart failure, weakened immunity and a tendency to recurrent infectious diseases.
However, it should be understood that not all patients experience obvious complications. In some cases, if you follow medical recommendations, constantly take medications and timely eliminate signs of ataxia, the quality of life of patients does not decrease and they manage to live to an old age.
Conservative therapy
In addition to drug therapy and surgical operations, complex therapy of the disease involves the appointment of other conservative methods. According to indications, patients may be prescribed speech therapy, occupational therapy, physiotherapy, and physical therapy. The latter involves both performing sports exercises and repeatedly repeating various household skills to improve coordination of movements: turning pages, pouring liquids, buttoning clothes.
Prevention of ataxia
In order to prevent the birth of people with ataxia, it is necessary to avoid the birth of children in families where there are patients with a hereditary form of this pathology. Disease prevention also includes:
- exclusion of consanguineous marriages,
- timely treatment of infectious diseases,
- blood pressure control,
- refusal to engage in sports that can lead to traumatic brain injury,
- compliance with the work and rest regime, rational nutrition.
This article is posted for educational purposes only and does not constitute scientific material or professional medical advice.