- Features of the disease and risk of transmission to offspring
- Symptoms of Gaucher disease
- First type
- Second type
- Third type
- Complications of sphingolipidosis
- Diagnostics
- General clinical and biochemical blood test
- Cell Enzyme Analysis
- Bone tissue and bone marrow examination
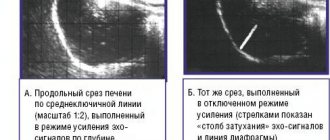
- Diagnostics of internal organs
- Genetic analysis
- Treatment of Gaucher disease
- Life expectancy with Gaucher disease
Gaucher disease is also called sphingolipidosis or glucosylceramide lipidosis.
This disease is hereditary and caused by genetic pathology. Gaucher disease was discovered as a separate disease in 1882 by the French physician Philippe Gaucher. He had the opportunity to treat a patient who had specific changes in the tissues of the spleen. The woman could not be saved due to blood poisoning. Several decades later, Mr. Gaucher encountered a similar clinical case. The specialist was able to determine that glucocerebroside accumulates in the tissues of the spleen and this process is accompanied by a deficiency of the enzyme glucocerebrosidase. Based on the examination results, the specialist suggested that these changes are associated with genetic pathology.
As a phenomenon, Gaucher disease is quite rare. Out of 70 thousand subjects, the mutation gene was found in only one person. It has been noted that in some peoples the prevalence of this disease is several tens of times higher. This is due to historical and cultural traditions that allow marriages between related people.
Features of the disease and risk of transmission to offspring
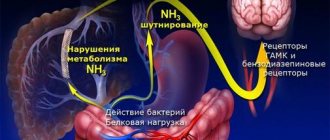
Gaucher disease belongs to the category of hereditary enzyme pathologies. Due to insufficient production of lysosomal enzymes, fat metabolism is disrupted. Lysosomes are cellular organelles - cell components that contain several dozen hydrolytic enzymes. They are necessary for the breakdown of proteins, fats, carbohydrates, and nucleic acids. Due to improper functioning of lysosomes, so-called storage diseases occur, in which lipids, mucopolysaccharides, and glycoproteins accumulate in cells. More than fifty hereditary storage diseases are known today.
In Gaucher disease, due to a deficiency of the enzyme beta-glucosidase, the breakdown of glycosphingolipids - compounds of lipids and carbohydrates - is impaired. Normally, the enzyme cleaves glucose from glucocerebroside. If this does not happen, macromolecules accumulate in cellular structures, forming so-called Gaucher cells. They are otherwise called foam cells because of their characteristic shape. When fat metabolism is disrupted, disorders in the functioning of various organs and systems begin. The liver, spleen, bone marrow, and musculoskeletal system are most affected.
The disease is inherited in an autosomal recessive manner. If both parents have a defective gene, the risk of having a child with enzyme pathology is 25%. The chance that a newborn will be a carrier of the damaged gene reaches 50% if only one parent or both have the mutation. For couples whose close relatives have Gaucher disease, medical genetic counseling is indicated. In the first trimester of pregnancy, amniotic fluid is analyzed to determine the level of enzyme activity in the fetus. If a pathology is detected, the woman is offered to terminate the pregnancy.
Causes
The disease is caused by changes (mutations) in the GBA gene.
All three forms of Gaucher disease are inherited in an autosomal recessive manner. Human traits, including classical genetic diseases, are the product of the interaction of two genes, one from the father and the other from the mother.
Recessive genetic disorders occur when a person inherits an abnormal gene from each parent. If a person receives one normal gene and one abnormal gene for a disease from each parent, the person will be a carrier of the disorder, but usually asymptomatic. The risk of two carrier parents both passing on the abnormal gene and therefore infecting the child is 25% in each pregnancy. The risk of having a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for men and women.
Symptoms of Gaucher disease
Symptoms of the disease are determined by the type of Gaucher disease. There are three types of pathology. Common to all types are an increase in the size of the liver and spleen, which interferes with their normal functioning, and disturbances in the hematopoietic system. Otherwise, the symptoms and age of onset of the pathology depend on the type of disease.
First type
This variety is the most common. Immediately after birth, the child begins to gradually enlarge the liver and spleen, but clinical manifestations of the pathology occur later. In some patients, the disease does not manifest itself until the age of 30–40.
Disorders of the hematopoietic system are manifested by anemia, decreased platelet levels, and bleeding. Patients complain of pain in bones and joints. Children may experience deformation changes, which disrupt the functioning of the musculoskeletal system. The disease also negatively affects the growth of the child: usually a child with Gaucher disease grows below average height. Due to the negative impact of the disease on bone tissue, patients with Gaucher disease are susceptible to osteoporosis and fractures.
In adult patients, the disease may manifest itself as increased skin pigmentation. Yellow and brown spots appear on the face and legs, and redness may occur in the area around the eyes.
Second type
It is rare and is characterized by manifestation in early childhood. As a rule, this occurs in the first year and a half of life.
As with the first type, enzyme pathology is manifested by an enlargement of the liver and spleen. The newborn's swallowing reflexes are impaired and breathing problems may occur. Gaucher cells form in the central nervous system, which is manifested by neurological disorders.
The child is noticeably lagging behind in physical and mental development. Up to a year, his muscle tone is reduced, and then hypertonicity occurs. It manifests itself most strongly in the neck and limbs, limits mobility, and causes pain. Hypertonicity leads to spasms and paralysis. Neurological manifestations include seizures. Many patients develop strabismus. Children with type 2 Gaucher disease are susceptible to pneumonia, which is severe.
The peculiarity of the pathology is that it is difficult to treat and leads to severe disability from childhood and early death of the patient.
Third type
The first symptoms of the disease appear at 2–3 years of age. During the examination, an enlargement of the liver and spleen is detected. In childhood or adolescence, damage to the central nervous system begins, which causes characteristic changes. The child experiences muscle spasms and convulsions, develops strabismus, muscle hypertonicity, and may experience problems with swallowing and breathing.
Cognitive functions gradually decline, the child loses the ability to speak and write. The disease manifests itself as emotional instability, even psychosis. Adolescents are delayed in sexual development.
The peculiarity of Gaucher disease of the third type is the rapid progression and deterioration of the patient's condition.
Classification and types of disease
The nature of the course of the disease varies in severity. Complications occur in childhood and adulthood. There are three types of disease:
- The first non-neuronopathic type. Sociology shows that it is common among Ashkenazi Jews. This pattern is called the Gaucher reaction. The clinical picture is characterized by a moderate, sometimes asymptomatic course of the disease. The psychology of behavior does not change, the brain and spinal cord are not damaged. Symptoms appear more often after thirty years of age. There are known cases of diagnosis in childhood. Timely treatment gives a favorable prognosis.
- The second type represents the neuronopathic infantile form and is rare. Symptoms appear in infancy as early as six months. Progressive damage to the child's brain occurs. Death can occur suddenly from suffocation. All children die before reaching two years of age.
- The third type (neuronopathic juvenile form). Symptoms have been observed since the age of 10 years. The intensification of symptoms is gradual. Hepatosplenomegaly - enlargement of the liver and spleen - is painless and does not impair liver function. Possible violation of behavioral psychology, the onset of neurological complications, portal hypertension, venous bleeding and death. Damage to bone tissue by Gaucher cells can lead to limited mobility and disability.
Complications of sphingolipidosis
Without treatment, patients may die due to serious problems in the liver and spleen, where Gaucher cells primarily accumulate. The most life-threatening are the second and third types of diseases, which affect the central nervous system. Patients experience changes in the structure of the spinal cord and brain. Spasm of the larynx and respiratory dysfunction can lead to acute oxygen deficiency and death from suffocation. Due to a decrease in platelet levels, the risk of bleeding increases, which due to its large scale is life-threatening. In the first type of sphingolipidosis, the musculoskeletal system is primarily affected. The patient's bone tissue is destroyed and its density is lost. In case of injury, this leads to serious fractures. Bone infections may also occur.
Due to spasms and paralysis, the patient has difficulty moving independently and requires constant assistance and care.
Description
Gaucher disease is a genetic pathology, which is based on a decrease in the functionality of the enzyme β-glucocerebrosidase, which ensures the breakdown of metabolic products in the cell.
The disease is a hereditary fermentopathy, a lysosomal storage disease. The code in the International Classification of Diseases 10th revision is E 75.2. Gaucher disease was named after the doctor who first described it. He identified the type of cells that are pathognomonic for this fermentopathy - macrophages with lipid accumulations. Later in the medical community they began to be called Gaucher cells.
The disease is considered rare, occurring in 1 person out of 50 thousand people. The exception is the population of Ashkenazi Jews, among them the prevalence of pathology is 1:450.
Inheritance of Gaucher disease is autosomal recessive. The basis is mutations of the gene that encodes the structure of the beta-glucocerebrosidase enzyme molecule. Its localization is region q21, the first chromosome. If both alleles are mutant, then the enzyme’s ability to break down metabolic products is reduced by 30%. As a result, macrophage lysosomes accumulate lipid molecules. This leads to chronic activation of the macrofogal system: the liver, spleen and bone marrow become enlarged due to the accumulation of pathological cells. Patients develop splenomegaly (enlarged spleen), hepatomegaly (enlarged liver), bone marrow infiltration, and damage to the osteoarticular system.
General clinical and biochemical blood test
Laboratory test results reveal a decrease in hemoglobin, leukocytes and platelets. Based on the results of biochemistry, it can be determined that glucocerebrosidase activity is reduced.
Cell Enzyme Analysis
Dried blood samples and skin fibroblasts are used as research material. In Gaucher disease, decreased activity of the enzyme glucosidase is found with a pronounced increase in the activity of an enzyme called chitotriosidase.
Bone tissue and bone marrow examination
To exclude oncological diseases of the hematopoietic system, the patient undergoes a bone marrow examination. In Gaucher disease, cellular structures characteristic of this pathology are found.
To assess the condition of bone tissue, densitometry, MRI, and x-rays are prescribed. The patient has osteopenia and osteoporosis, areas of osteonecrosis.
Diagnostics of internal organs
The very first marker of Gaucher disease is an enlargement of the liver and spleen. To detect pathology, an abdominal ultrasound or MRI is performed.
Genetic analysis
If, based on the results of a comprehensive diagnosis, it is not possible to accurately determine the presence of a pathology, a special DNA test is prescribed that will confirm or deny the presence of a defective gene.
Diagnostics
The diagnosis of Gaucher disease should be considered in persons with unexplained anemia and mild bruising, especially if they have an enlarged spleen and liver and fractures. Diagnosis of the disease can be confirmed by careful clinical assessment and various specialized tests, particularly tests (eg, enzyme assays) that measure acid beta-glucosidase activity in white blood cells or skin cells (fibroblasts) and genetic (DNA) analysis of causative gene defects (mutations) . Note: The enzyme test cannot reliably detect carriers.
An enzyme assay test is a standard tool used by doctors to diagnose someone who is thought to have Gaucher disease because these patients typically have low glucocerebrosidase enzyme activity. If the results are slightly low, then the doctor should refer the person for genetic testing for mutations in the GBA gene. Genetic testing is done through blood or saliva. Identification of two causative gene defects in combination with the results of enzyme tests confirms the diagnosis of the disease. Individuals who have only one gene defect may be carriers or, if low beta-glucosidase is present, may be affected by a second gene defect (mutation) that has not been detected. In this situation, the patient can be referred to an appropriate genetic specialist. DNA testing identifies people who carry a mutation in the GBA gene, who can pass the mutation on to their children.
Prenatal diagnosis of Gaucher disease is possible if a known GBA gene mutation is present in the family. Testing can be done with amniocentesis or chorionic villus sampling (CVS), but this is rare unless there is a family history of Gaucher disease type 2. During amniocentesis, a sample of the fluid that surrounds the fetus (amniotic fluid) is removed and analyzed, while CVS involves removing tissue samples from part of the placenta. A prenatal diagnosis can confirm a specific diagnosis of the disease, but does not determine the type.
Treatment of Gaucher disease
Therapy for Gaucher disease is primarily aimed at replenishing enzyme deficiency. The patient constantly takes drugs based on recombinant glucocerebrosidase, which is administered by intravenous injection. This treatment gives a positive effect in the first and third types of the disease. In some patients, the progression of the disease can be stopped and they can lead a normal life.
In the USA and European countries, people with moderate manifestations of the disease are prescribed a substrate - reducing therapy. Glucosylceramide synthase inhibitors are used as drugs. Their use reduces the formation of glycosphingolipid substrates and slows down their accumulation by accelerating the catabolism of macromolecules.
Treatment for Gaucher disease also includes drug therapy that slows bone destruction. First of all, these are calcium and vitamin D preparations, foods rich in this mineral. Antibacterial therapy is indicated for infectious bone lesions. Non-steroidal anti-inflammatory drugs are prescribed to relieve pain.
To reduce neurological symptoms, muscle relaxants and nootropic drugs are used.
In the second type of Gaucher disease, it is necessary to monitor respiratory activity and promptly eliminate failures that can lead to respiratory arrest.
Some patients with sphingolipidosis undergo surgical removal of the spleen or bone marrow transplant.
Forecast
With the first type of disease, early diagnosis, and timely initiation of replacement therapy for Gaucher disease, positive dynamics are possible. The second type of glucocerebrosidosis is the most unfavorable, as it is more severe. Sick children usually do not live more than two years. The third form of Gaucher disease, with timely diagnosis and adequate treatment, allows maintaining the patient’s vital functions. Otherwise, he dies quite quickly from complications that develop.
Fatty liver hepatosis: symptoms and treatment Liver diseases: symptoms and first signs Hepatitis C: first signs and treatment regimen Why is the liver enlarged - what to do and how to treat? Malabsorption syndrome: symptoms and treatment Liver cirrhosis
Life expectancy with Gaucher disease
The most favorable outcome is typical for the first type of disease. It appears late and is more treatable than others, allowing the patient to live into adulthood. With the third type, the life expectancy of most patients was previously 15–17 years, but thanks to improvements in treatment, many manage to live up to 30–40 years.
The most unfavorable prognosis is typical for the second type. Pathological changes appear in the first months of life, and the disease progresses rapidly. Most patients die within the first two years of life.
Affected Populations
All forms of Gaucher disease affect men and women in equal numbers. Type 1 disease is the most common type, accounting for more than 90 percent of cases among Caucasians. Patients with type 1 disorder typically begin to experience symptoms during adolescence, but the age of onset varies from childhood to adulthood. The age of onset of type 2 Gaucher disease is early childhood. The age of onset of Gaucher disease type 3 varies, but the disorder usually begins in childhood or adolescence. The frequency of neuropathic forms of Gaucher disease, that is, the proportion of such cases, is higher among non-Caucasians.
There are approximately 6,000 people in the United States with Gaucher disease. Unknown in Russia. The disorder is the most common genetic disorder in people of Ashkenazi Jewish descent, where incidence can be as high as 1 in 450 births. There is no ethnic prevalence associated with Gaucher disease type 2 or 3. However, there is a subtype of Gaucher disease type 3 that occurs with greater frequency in the Norrbotten region of Sweden (Norrbotten Gaucher disease). The estimated prevalence in the Swedish population of Norrbotten is 1 in 50,000 people.
Sources
- Clinical guidelines for providing medical care to children with Gaucher disease. /Gundobina O.S., Savostyanov K.V., Pushkov A.A., Belogurova M.B., Bukina T.M., Zakharova E.Yu., Mikhailova S.V., Novikov P.V. – 2014
- National clinical guidelines “Diagnostics and treatment of Gaucher disease.” /Lukina E.A., Sysoeva E.P., Mamonov V.E., Yatsyk G.A., Tsvetaeva N.V., Gundobina O.S., Finogenova N.A., Smetanina N.S., Novikov P.V. – 2014.
- Gaucher disease in children. / Basistova A. A., Baranov A. A., Kaganov B. S., Bukanovich O. V. - 2004.
- Gaucher disease in children. /Gundobina O.S., Komarova E.V., Namazova-Baranova L.S., Gevorkyan A.K., Movsisyan G.B. - 2013.
- Gaucher disease: modern diagnosis and treatment. /Lukina E. A. – 2009.
- Lysosomal storage diseases: Gaucher disease. /Davydova A.V. – 2009.
Medicines
Photo: unsplash.com
Several drugs for enzyme replacement treatment are registered in Russia. One is produced using cell lines derived from human fibroblasts, the other from cell lines cultured from Chinese hamster ovaries. The amino acid sequence of these drugs is similar to that of natural beta-glucocerebrosidase, but has longer glycan chains and some other structural differences that facilitate absorption by macrophages.
Symptomatic drug therapy for Gaucher disease is aimed at reducing the manifestations of osteoporosis. Patients are prescribed bisphosphonates, alfacalcidol, and calcium salts. Thanks to these drugs, bone loss slows down or completely stops, bone strength increases, and fractures are prevented. Analgesics are used to reduce pain.
Folk remedies
Gaucher disease requires highly qualified medical care and the use of modern medications for enzyme replacement treatment. Traditional methods for this disease are ineffective, since the pathological processes are caused by a genetic mutation and enzyme deficiency.
The information is for reference only and is not a guide to action. Do not self-medicate. At the first symptoms of the disease, consult a doctor.
SEARCH FOR TREATMENT AROUND THE WORLD WITH YELLMED