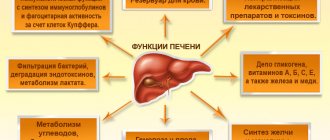
Hepatic encephalopathy (HE) is a reversible change in the neuropsychic sphere, manifested by lethargy, decreased concentration, cognitive dysfunction, sleep inversion, intentional tremor of the limbs, caused by metabolic disorders in liver cirrhosis (LC). PE is a consequence of hepatic cell failure, which develops in almost all known liver diseases (viral and toxic hepatitis, autoimmune liver disease, Wilson's disease, etc.), is associated with a decrease in the volume of functioning liver parenchyma and the inability of hepatocytes to maintain homeostasis and perform a detoxifying function [2, 3 , 5].
The extreme degree of HE is hepatic coma with the development of irreversible structural changes in the nervous system, which are often preceded by a generalized convulsive syndrome. Hepatic coma is the result of long-term HE or fulminant hepatic process, which often ends in death [3].
Mechanism of development of PE
The pathogenesis of PE is still under study. There are several concepts for the development of this syndrome.
1. The most substantiated theory is the disruption of energy processes in neurons, according to which non-ionized ammonia easily penetrates the blood-brain barrier (BBB) and neuronal membranes. In this case, the rate of glucose oxidation decreases, which leads to energy starvation of brain cells [2].
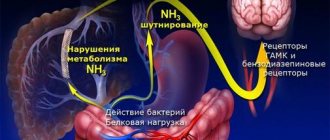
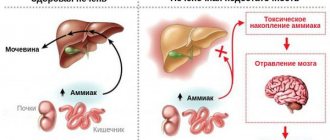
Ammonia, normally produced in the colon, travels through the portal vein to the liver, where most of it is included in the ornithine cycle, the end product of which is urea. Urea synthesized in the liver enters the blood, then is filtered into the lumen of the proximal tubules of the kidneys and is ultimately excreted in the urine. Ammonia that is not included in the ornithine cycle is captured by a small population of perivenous hepatocytes, where glutamine is formed under the influence of glutamine synthetase. These mechanisms help prevent toxic products from entering the systemic circulation. With liver cell failure, there is a decrease in the rate of metabolism of ammonia and other toxins in the liver. In addition, toxic products can enter the general bloodstream, bypassing the liver, through portocaval anastomoses. The level of hyperammonemia in 90% of patients with cirrhosis significantly correlates with the development and severity of PE.
Thus, ammonia derivatives can enter the brain tissue from the blood through the BBB and have a pronounced toxic effect on the central nervous system, the mechanism of which is currently under active study.
2. According to the theory of (false) neurotransmitters, in patients with liver diseases, with increased protein catabolism, a significant amount of aromatic amino acids (tyrosine, phenylalanine, tryptaphan) enters the blood and at the same time the content of branched-chain amino acids (valine, leucine, isoleucine) decreases. Amino acid imbalance leads to the penetration of a mass of aromatic amino acids through the BBB, which contributes to a decrease in the synthesis of dopamine and the formation of false neurotransmitters (octopamine, phenylethanolamine). The enzyme system is inhibited and norepinephrine is formed, impulse transmission in the catecholamine and dopamine synapses of the brain is disrupted, which leads to depression of the nervous system, brain exhaustion and the development of encephalopathy [1, 3].
In addition, a decrease in the inactivating function of the liver leads to an imbalance between the synthesis and breakdown of γ-aminobutyric acid (GABA), which is the main inhibitory neurotransmitter in the brain. This hypothesis is confirmed by the presence of high levels of GABA and GABA-like substances in the blood plasma and cerebrospinal fluid in patients with PE [1].
3. Bacterial overgrowth syndrome leads patients with cirrhosis to the development of endotoxemia, translocation of decay products of intestinal bacteria and their toxins into the portal vein system with the formation of systemic manifestations, including PE. There are several reasons that contribute to the excessive growth of gram-negative and anaerobic intestinal microflora and endotoxemia in cirrhosis: the syndrome of impaired digestion and absorption, associated changes in the composition of microorganisms, hypomotility of the small and large intestine, increased permeability of the intestinal wall. In addition, changes in the composition of the intestinal flora are associated with an increase in the number of intraepithelial lymphocytes, a decrease in their proliferative activity and the ability to produce interferon, which leads to impaired local immunity [4, 5, 9].
Based on all of the above, we can conclude that with PE, metabolic processes in brain cells are disrupted, but today there is no single explanation for the development of this pathological condition. External factors contributing to the development of PE in cirrhosis are given in Table. 1.
Description and causes of encephalopathy
Hepatic encephalopathy is a serious disease
The term “encephalopathy” in medicine refers to a disorder of brain activity as a result of non-inflammatory processes.
In this case, dysfunction appears due to dystrophic changes in the structure in the tissues and cells of the brain.
One of the factors causing this pathology is serious liver diseases:
- cirrhosis;
- cancer;
- alcoholic hepatitis;
- acute viral hepatitis.
All these diseases cause acute liver failure. In addition, there are other causes of hepatic encephalopathy that develops as a result of infection of the blood with intestinal neurotoxins:
- excess protein in the body due to poor nutrition;
- drug intoxication;
- alcohol abuse;
- bleeding in the stomach, intestines;
- surgical intervention;
- frequent constipation;
- ascites (accumulation of fluid in the peritoneum) and subsequent peritonitis (acute inflammation).
Liver failure is accompanied by a number of serious disorders in the body. Hemostasis, hydrostatic and oncotic pressure, water-electrolyte and acid-base composition of the blood change. These changes affect astrocyte cells, which make up one third of the brain.
Normally, they should ensure sufficient permeability of brain tissue to blood, neutralize toxins, and supply cells with electrolytes and neurotransmitters.
In hepatic encephalopathy, the central nervous system is affected. Astrocytes lose their functions under the influence of ammonia, fatty acids, magnesium, false transmitters, amino acids, as a result of which intracranial pressure increases and cerebral edema develops.
Clinical picture
Currently, there are three clinical variants of PE [2, 3]:
1. Subclinical encephalopathy
Subclinical PE is characterized by the absence of clinical symptoms, but additional studies reveal a number of neuropsychic disorders (deterioration of cognitive abilities, loss of fine motor skills, which can only be determined using psychometric tests). According to the literature, in patients with cirrhosis, subclinical PE is detected in 50–70% of cases. In patients with cirrhosis with portosystemic shunting, PE can be episodic with spontaneous resolution or intermittent, lasting many months or even years.
2. Chronic encephalopathy
This form of PE is divided into three stages (Table 2). In stage I, subtle personality changes, absent-mindedness, decreased attention, inability to concentrate, mild ataxia, and sleep disturbances are clinically observed.
Stage II is manifested by fatigue, drowsiness, apathy, monotonous speech, inappropriate behavior with noticeable changes in the “structure” of the personality, disturbances in time orientation, the presence of intention tremor, reflexes of oral automatism, and ataxia.
Stage III PE is characterized by severe disorientation in time and space, incoherent speech, aggression, hypo- or hyperreflexia, pathological reflexes (Gordon, Zhukovsky), pronounced intention tremor, and muscle rigidity. With inadequate treatment of PE, hepatic coma may occur, which is characterized by a lack of consciousness and disappearance of reactions to painful stimuli. Rigidity of the muscles of the limbs and the back of the head, a mask-like face, and pathological reflexes (Babinsky, Gordon, Zhukovsky) are noted. Sometimes grasping and proboscis reflexes are detected [5].
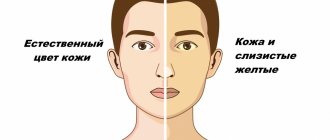
In addition to psychoneurological symptoms, coma is characterized by clinical manifestations of hepatic cellular failure - jaundice, liver odor, ascites, hemorrhagic symptoms. The causes of death are cerebral edema, pulmonary edema, hepatorenal syndrome, infectious-toxic or hypovolemic shock.
3. Acute PE
It can develop in patients with severe jaundice against the background of ascites with severe necrotic processes in the liver, as well as in patients with fulminant liver failure. Acute HE may be accompanied by the development of hepatic coma.
Diagnostic procedures
For diagnosis, it is enough to take a blood test and do an ultrasound.
The diagnosis of hepatic encephalopathy includes the establishment of clinical symptoms, assessment of the stage of development and severity of the disease.
First of all, significant attention should be paid to collecting anamnesis - abuse of medications and alcohol, acute viral hepatitis or surgery, the presence of constipation.
Next, the patient must pass:
- General blood analysis. The presence of pathology is indicated by anemia, low platelet concentration, an increase in the content of blood leukocytes (leukocytosis) with toxic granularity of neutrophils.
- Liver tests. This reveals an increase in bilirubin and transaminase activity, and the activity of liver enzymes - gamma-glutamyltransferase and alkaline phosphatase.
- Coagulogram. In this case, there is a change in hemostasis, a decrease in fibrinogen and an increase in thromboplastin.
If necessary, other laboratory tests are prescribed that will help identify multiple organ failure.
Clarification of the diagnosis and assessment of the severity of hepatic encephalopathy is carried out based on the following procedures:
- Liver biopsy using puncture method.
- Magnetic resonance imaging of the liver and biliary tract.
- Ultrasound examination of the liver and gall bladder.
- Computed tomography of the biliary tract.
- Electroencephalography.
In order to identify the influence of diseases of other organs of the patient on the development of encephalopathy, differential diagnosis is carried out.
These studies are used in cases of metabolic disorders, infectious diseases of the central nervous system, hemorrhagic stroke, cerebral hemorrhage, increased nitrogen levels in the blood due to abuse of alcohol or medications.
Diagnostics
Diagnosis of PE is carried out on the basis of anamnesis, patient complaints, corresponding symptoms (Table 2) and central nervous system disorders characteristic of cirrhosis of various etiologies identified during clinical and instrumental research.
The simplest and most objective in clinical practice is the use of psychometric tests [2, 5, 7]: Number connection test: when performing this test, the patient’s task is to connect a group of numbers from 1 to 25 as quickly as possible. A time exceeding 30 seconds is considered pathological.
Line test: The essence of the line test is for the patient to draw a line in a corridor limited on both sides, without touching the outer boundaries. The obtained parameters are compared with normal values for a given age group.
These tests make it possible to diagnose, among other things, subclinical PE. There is not a single pathognomonic feature for PE; the totality of clinical data and the doctor’s experience have diagnostic significance.
It is important to resolve the issue of the etiology of the liver disease, which underlies PE, since only qualified tactics of a hepatologist can provide a favorable prognosis for the disease.
Symptoms
Hepatic encephalopathy occurs in several stages, during which the severity of clinical manifestations varies. In general, the disease is accompanied by a wide range of mental and neurological disorders. The main ones are described below.
Sleep disorders
. Patients with hepatic encephalopathy initially experience drowsiness. Subsequently, an inversion (radical change) of the normal rhythm of wakefulness and sleep occurs.
Impaired consciousness
. With this disease, there is usually a decrease in the number of spontaneous actions. The patient rarely fixes his gaze, he exhibits apathy and lethargy, and answers questions briefly. As the condition worsens further, a person suffering from hepatic encephalopathy responds only to intense stimuli.
Personality changes
. Patients may experience a loss of interest in the family and childishness. Moreover, these changes often persist even during the period of remission, which indicates the involvement of the frontal lobes of the brain in the pathological process. With hepatic encephalopathy, patients may be in a state of euphoria or a playful mood.
Intellectual disorders
. Impairments in cognitive functions (memory, attention, etc.) can be mild or severe. In severe cases, confusion occurs.
Speech disorders
. In patients with hepatic encephalopathy, the voice becomes monotonous, and speech becomes slurred and slow. In the future, speech impairment may become even more severe (dysphasia).
Flapping tremor
. With this disease, a characteristic flapping (flexion-extension) movement of the hand is observed with outstretched arms and widely spaced fingers.
Coma
. At first it may resemble a normal dream, but later the patient stops responding to external stimuli completely.
Pathogenesis of hepatic encephalopathy: basic principles
Traditionally, PE is divided into overt (with neurological and/or psychiatric abnormalities that can be detected clinically) and latent (with abnormalities that are detected using neuropsychological or neurophysiological tests).
It is believed that the following main factors underlie the pathogenesis of PE: 1) increased ammonia levels in the blood (hyperammonemia) due to the excessive entry into the blood of nitrogen-containing substances synthesized by intestinal bacteria and insufficient neutralizing function of hepatocytes, 2) portosystemic shunting of the blood. A serious role in the pathogenesis of PE is played by the severity of liver damage, the severity of inflammation and/or oxidative stress, age, associated diseases (diabetes mellitus, renal failure), the severity and nature of the factor provoking PE.
Astrocytes absorb ammonia, which is involved in the reaction of converting glutamate into glutamine; an excess of the latter in hyperammonemia leads to swelling of astrocytes and tissue edema. A decrease in glutamate content in astrocytes is accompanied by a disruption of its neurotransmission with the participation of NMDA receptors, as well as an increase in the activity of GABAergic processes. Most likely, these events underlie cognitive deficits, convulsive paroxysms and other clinical manifestations of PE [5, 6].
Despite the long period of studying PE, there are still issues that require clarification: correct diagnosis of both clinically pronounced and latent PE; the role of certain factors in its pathogenesis and, accordingly, diagnostic tools, the choice of optimal therapeutic measures. Separate issues: tactics for managing patients with minimal PE and outpatient treatment, namely monitoring the level of consciousness, the effectiveness of treatment for PE at home.
Introduction
Hepatic encephalopathy (HE) remains one of the most serious complications of liver cirrhosis. The variety of its clinical manifestations creates certain difficulties in optimally fast and correct diagnosis right at the patient’s bedside. In the international community, clinical guidelines serve as the main document guiding the activities of doctors in everyday practice. Regarding PE, they were created by the American and European Associations for the Study of the Liver [1]. The provisions of this document are taken into account in Russian recommendations for the management of patients with liver cirrhosis [2]. However, many years have passed since they were written, and new knowledge and experience have been accumulated. This new knowledge and provisions are summarized in several works aimed at optimizing clinical work with patients suffering from PE: 1) consensus of the International Society for the Study of PE and Nitrogen Metabolism (ISHEN: The International Society for Hepatic Encephalopathy and Nitrogen Metabolism) from 2021. [3]; 2) a review article by world experts in the field of PE “A new look at the classification, pathophysiology and therapy of PE” [4]; expert consensus statements on the management of patients with decompensated cirrhosis prepared by the European Association for the Study of the Liver and published in J. Hepatology
in 2021 [5]. They form the basis of new updated recommendations for the management of patients with cirrhosis of the liver of the Russian Society for the Study of the Liver, which are now being prepared for publication and adoption by the Ministry of Health of the Russian Federation.
This review presents new knowledge and provisions of recent years on the issue of clinical work with patients suffering from PE due to liver cirrhosis.
Principles of treatment of hepatic encephalopathy
The dominant role of ammonia in the pathogenesis of HE in liver cirrhosis determines the main direction of treatment - reducing its concentration, which is achieved by reducing its production in the intestine and increasing elimination by hepatocytes. Accordingly, in order to reduce the production of ammonia in the intestine, the following main drugs are used: the non-absorbable antibiotic rifaximin and the osmotic laxative lactulose (lactitol). L-ornithine-L-aspartate is used to stimulate ammonia clearance by hepatocytes (through urea formation and glutamine synthesis) and/or muscle cells. Detailed regimens for prescribing these drugs are indicated in the clinical recommendations of the Russian Society for the Study of the Liver for the treatment of patients with cirrhosis of the liver in the section “treatment of hepatic encephalopathy” [2]. In contrast to European and American recommendations, in the Russian Federation, any of the above drugs can be used as first-line therapy at the discretion of the attending physician and analysis of the characteristics of the disease in a particular patient.
Lactulose (lactitol) is a laxative that has little effect on the composition or function of the intestinal microbiota, its effect probably being realized by accelerating intestinal transit and acidifying the intestinal environment. As a result, ammonia production in the intestine decreases, and its excretion in feces increases. A large number of randomized clinical trials, as well as observational studies, have shown the benefit of lactulose in the treatment of PE compared with no therapy, although there are no true double-blind studies because It is very difficult to “blind” the laxative effect and typical sweet taste of this drug. The clinical use of lactulose is limited by its side effects: diarrhea, nausea and bloating. Diarrhea can lead to electrolyte imbalance and even worsen PE. Accordingly, lactulose intake should be started with 15–20 ml every 12 hours and gradually increased individually to achieve the goal: soft stools 2 times a day.
Antimicrobial agents. Rifaximin is a non-absorbable antibiotic that is administered orally and does not lead to the development of bacterial resistance. Rifaximin has a positive effect on the microbial composition of the intestine, which leads to a decrease in ammonia production. The drug has a good safety profile, can be used long-term and does not require dose adjustment in patients with liver and/or renal failure. Treatment with rifaximin leads to resolution of PE and reduces mortality from this complication. The combination of rifaximin with lactulose is more effective than the use of lactulose as monotherapy [4].
L-ornithine-L-aspartate (LOLA) has been studied in a large number of randomized clinical trials. In 2021, a review of randomized clinical trials and meta-analyses was published, which analyzed the effectiveness of LOLA in patients with cirrhosis in comparison with placebo, the absence of any interventions, and other drugs for the treatment of PE (lactulose, rifaximin, etc.). Intravenous administration of LOLA at a dose of 20–40 mg per day for 7 days improved the mental status of patients with cirrhosis classes A and B on the Child–Pugh scale by 3.22 times, compared with placebo, reduced the manifestations of PE and reduced postprandial ammonia levels . This property of the intravenous form of the drug can be successfully used in the differential diagnosis of HE in liver cirrhosis with other conditions that affect the mental status of patients, such as alcohol withdrawal syndrome. This is especially true in intensive care units. In this situation, the correct diagnosis is based on the “ex juvantibus” principle, since hyperammonemia is a key factor in the pathogenesis of PE, and not the withdrawal syndrome.
Pooled analysis of all studies demonstrates the effect of both intravenous and oral LOLA on both symptomatic and silent HE and on blood ammonia levels compared with placebo/no treatment. LOLA is not inferior in treatment effectiveness to the non-absorbable antibiotic rifaximin and the osmotic laxative lactulose [11].
The treatment program for complications of liver cirrhosis includes human albumin; its ability to remove toxic compounds from the circulation is relevant in the treatment of PE.
Hepatic encephalopathy (HE), or portosystemic encephalopathy, is a potentially reversible syndrome of brain dysfunction in patients with progressive liver failure. However, P.E. is not a single category and may reflect the clinical manifestations of reversible metabolic encephalopathy, brain atrophy due to hepatocerebral dystrophy, cerebral edema, or any combination of these conditions. The mechanisms that impair brain function in liver failure are still not fully understood, but it is clear that they are directly related to liver failure and impaired ammonia metabolism. If the underlying liver disease is not curable, PE is associated with poor survival and a high risk of relapse [1, 2]. Even in its mildest form, PE reduces health-related quality of life and is a risk factor for episodes of severe PE [3, 4].
Pathogenesis
Despite more than 100 years of research, the pathogenesis of PE is still not entirely clear. This is mainly due to the difficulty of studying the brains of patients with PE in vivo
. Most published data come from experimental models of PE, which are far from perfect. The most common hypotheses for pathogenesis reflect the role of neurotoxins, impaired neurotransmission due to metabolic disturbances in liver failure, changes in brain energy metabolism, amino acid imbalance, systemic inflammatory response and disturbances in the permeability of the blood-brain barrier [5–7]. Various hypotheses for the pathogenesis of PE are not mutually exclusive. It is likely that many of the described disorders may act simultaneously and ultimately lead to the development of PE.
Neurotoxins.
The most studied neurotoxin associated with HE is ammonia, which is produced primarily in the gastrointestinal tract and travels through the portal vein to the liver. A healthy liver detoxifies incoming ammonia by converting it into glutamine, thereby preventing it from entering the systemic circulation. With progressive liver diseases, an increase in the concentration of ammonia in the blood occurs both as a result of a violation of its conversion by the liver into glutamine, and as a result of porto-systemic shunting, in which blood, bypassing the liver, enters the systemic circulation [3].
When the concentration of ammonia in the blood increases, it penetrates the blood-brain barrier and has a neurotoxic effect primarily on astrocytes, the most numerous cells in the brain, closely associated with the functioning of neurons. A key mechanism for the development of PE is astrocyte swelling due to hyperammonemia [8–11].
Excess ammonia leads to the accumulation of glutamine formed in astrocytes, which is accompanied by an increase in intracellular osmolarity and in high concentrations causes brain edema [12].
Impaired neurotransmission.
Impaired functioning of cerebral cortex neurotransmitters and their receptors plays an important role in the pathogenesis of PE. Abnormalities in several neurotransmitter systems have been studied in experimental models of liver failure. Most of these studies describe changes in the GABAergic neurotransmitter system, dopaminergic, serotonergic and glutamatergic neurotransmitter systems [13, 14]. In particular, the role of GABAergic influences in the development of PE may be associated with the activation of benzodiazepine receptors in the brain and the associated increase in the synthesis of neurosteroids [15].
Amino acid imbalance.
An important role in the pathogenesis of PE is played by amino acid imbalance in the form of an increase in the level of aromatic amino acids (tyrosine, phenylalanine, tryptophan), which are precursors of false neurotransmitters, and a decrease in the content of branched-chain amino acids (valine, leucine, isoleucine). Under these conditions, there is an excessive supply of aromatic amino acids to the brain, which serve as the starting product for the synthesis of false neurotransmitters. These changes in amino acid composition are also accompanied by a decrease in dopamine synthesis, which also contributes to the formation of false neurotransmitters [16].
Clinic
The clinical picture of PE is a wide range of nonspecific neurological and mental disorders [17]. Signs of encephalopathy in patients with chronic liver diseases depend on the etiology of the underlying disease, the nature and severity of pathogenic factors.
The severity of PE varies from latent (stage 0) and mild (stage I) to coma (stage IV). With minimal PE, it is manifested mainly by disturbances in abstract thinking and a general mild decline in cognitive functions without impairments of memory, intelligence, speech, and learning ability, which remain intact for a long time [18, 19]. In some patients with liver failure, for a number of years only disorders of higher brain functions are detected without any other neurological symptoms [20].
As PE progresses, personality changes increase, such as apathy, irritability and decreased behavioral control, as well as obvious changes in consciousness and motor function [21]. Sleep-wake cycle disturbances with excessive daytime sleepiness are common, although complete sleep-wake cycle disruption is usually absent [22–24]. With increasing liver failure, progressive disorientation in time and space, behavioral disturbances develop, episodes of confusion with agitation or drowsiness, stupor and, finally, coma occur [25]. The consensus of the International Society for Hepatic Encephalopathy and Nitrogen Metabolism (ISHEN) considers the appearance of disorientation or asterixis (“flapping” tremor) as the initial signs of overt HE [26].
In noncomatose patients with PE, motor dysfunction such as muscle hypertonia, hyperreflexia, and a positive Babinski reflex is observed. As coma progresses, deep tendon reflexes usually decrease [27] and even disappear, although pyramidal signs may persist. Occasionally, focal neurological deficits may occur [28]. Convulsions in PE are very rare [29–31].
Frequent manifestations of the disease are extrapyramidal disorders in the form of facial hypomia, muscle rigidity, bradykinesia, hypokinesia, monotony and slow speech, Parkinson-like tremor and dyskinesia with limitation of voluntary movements [27].
Asterixis (“flapping tremor”) is often noted in the early and middle stages of PE, which precedes stupor or coma. It is not actually a tremor but a negative myoclonus and is caused by hyperextension of the patient's wrist with the fingers spread apart. It is noteworthy that mental and motor disorders in PE may be mild or do not progress equally in different patients, which leads to difficulties in determining the stage of PE.
Classification
According to the practical recommendations of the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver [32], PE is classified according to four criteria: depending on the reasons that led to its development; by duration and nature of the flow; according to the severity of the course (stages) and depending on the presence of provoking (trigger) factors.
A - hepatic encephalopathy as a result of acute liver failure;
B — portosystemic shunting in the absence of liver cirrhosis (LC);
C - hepatic encephalopathy in patients suffering from cirrhosis.
Classification of PE by duration and nature of the course:
1) episodic PE;
2) recurrent PE - these are attacks of PE that occur with a time interval of 6 months or less;
3) persistent PE represents behavioral disturbances that are constantly present and interspersed with relapses of overt PE.
Classification of the severity of PE by stages (West Haven scale)
I. Minimal (latent) stage: there are no clear clinical symptoms, but there is a violation of standardized psychomotor tests (number connection test, line test).
II. The first (mild) stage: apathy, excitement, irritability, anxiety, euphoria, fatigue, disturbances in the rhythm of sleep and wakefulness, mild tremor, impaired coordination of movements, asterixis.
III. Second (middle) stage: drowsiness, lethargy, disorientation in time and space, inappropriate behavior, asterixis, dysarthria, ataxia.
IV. Third (severe) stage: stupor, severe disorientation, unclear speech, hyperreflexia, the presence of pathological reflexes (Gordon, Zhukovsky), myoclonus, hyperventilation.
V. Stage four: hepatic coma, decerebrate rigidity, oculocephalic phenomenon, lack of response to any stimuli.
Depending on the presence of provoking (trigger) factors, PE is divided into unprovoked and provoked (in this case, the provoking factor must be indicated). It is advisable to identify precipitating factors (infections, gastrointestinal bleeding, diuretic overdose, electrolyte disturbances, constipation) for all episodes of type C HE and eliminate them if present.
Diagnostics
Diagnosis of obvious PE
The diagnosis of overt PE is based on an objective clinical examination. It involves assessing the symptoms of HE, as well as excluding other causes of brain dysfunction. In the clinical picture, patients with obvious PE have symptoms of diseases of the hepatobiliary system, for example, in most patients the development of PE is preceded by jaundice. A liver odor and hyperventilation can often be detected in patients with encephalopathy [33]. Also, the diagnosis of PE is confirmed by identifying provoking factors (infections, bleeding and constipation, etc.).
The “gold standard” for diagnosing overt PE is the West-Haven criteria, however, they have limited diagnostic value due to the subjectivity of the assessments, especially in stage I PE, since mild hypokinesia, psychomotor retardation and absent-mindedness can be missed during clinical examination. The leading diagnostic symptoms of obvious PE are the presence of disorientation and asterixis in the patient [32].
To describe the condition of patients with significantly altered consciousness, it is recommended to use the Glasgow Coma Scale (GCS). The Glasgow scale involves calculating points that reflect the severity of the patient's reaction to stimuli. The ability to open the eyes, the nature of motor and verbal reactions to simple stimuli (voice and pain) are determined. Coma is preceded by less profound forms of depression of consciousness: confusion, stupor and stupor.
Diagnosis of minimal PE
Minimal HE is defined as abnormal brain function detected by tests in patients with chronic liver disease who are not confused or have not developed asterixis. The examination of such patients may include two main types of tests: psychometric and neurophysiological. ISHEN recommends the use of at least two tests, depending on their availability and local conditions. The importance of testing for minimal HE is that it predicts the development of overt HE.
To diagnose minimal PE, it is recommended to carry out neurophysiological and psychometric tests, among which the most important are both simple ones performed on paper with a pen (psychometric scale of PE) and computerized ones (reaction time delay test, Stroop test, inhibitory control test and SCAN test ) and neurophysiological (critical flicker fusion frequency test) tests [32]. Electroencephalography can detect changes in cortical brain activity in PE, but this method is not specific enough, since it can be influenced by concomitant metabolic disorders and medications.
Laboratory tests.
In patients with PE, serum-biochemical liver syndromes of varying severity are detected, depending on the predominant direction of pathological processes in the liver [34].
Cytolysis syndrome, or hepatocyte damage syndrome (violation of integrity, necrosis of hepatocytes), is characterized primarily by an increase in aminotransferases (aspartate and alanine aminotransferase), as well as other enzymes - lactate dehydrogenase-5, aldolase, ornithine carbamyltransferase, sorbitol dehydrogenase, glutamate dehydrogenase.
Cholestasis syndrome is characterized by an increase in bilirubin, mainly conjugated (direct), alkaline phosphatase, gamma-glutamyl transpeptidase, 5-nucleotidase, cholesterol and bile acids.
Inflammation syndrome is characterized by increased levels of various globulin fractions, dysproteinemia, and increased levels of serum immunoglobulins.
The AASLD/EASL guidelines [32] note that in patients with chronic liver disease, only high blood ammonia levels have diagnostic or prognostic value. However, if ammonia levels are within normal limits, the diagnosis of PE is questionable. Repeated ammonia measurements may be used to assess the effectiveness of treatment when patients are taking medications that lower ammonia levels.
Computed tomography or magnetic resonance imaging and other imaging methods do not provide complete diagnostic information and are of limited value. They are mainly used to exclude structural damage to the brain in patients with cirrhosis.
Differential diagnosis
AASLD/EASL guidelines indicate the need to exclude other diseases that may resemble PE [32]:
1) diabetes mellitus (hypoglycemic, hyperglycemic and hyperosmolar coma, lactic acidosis);
2) alcohol abuse (intoxication, withdrawal syndrome, Wernicke syndrome);
3) overdose of drugs (benzodiazepines, antipsychotics, opioids);
4) neuroinfections and electrolyte disorders (hyponatremia, hypercalcemia);
5) non-convulsive epilepsy, mental illness;
6) intracerebral hemorrhage and ischemic stroke;
7) dementia (primary and secondary);
 brain damage (traumatic, neoplastic, normal pressure hydrocephalus, obstructive sleep apnea syndrome).
brain damage (traumatic, neoplastic, normal pressure hydrocephalus, obstructive sleep apnea syndrome).
Treatment
Treatment of PE includes elimination of precipitating factors, dietary measures and drug therapy. Elimination of precipitating factors is the primary goal in the treatment of overt PE, since in 90% of patients this is sufficient to improve the condition [32].
Diet
Correction of disorders of protein-nitrogen metabolism is crucial in the treatment of all degrees of PE, since 75% of patients with PE have moderate or severe protein-calorie deficiency, accompanied by loss of muscle mass. Long-term restriction of protein intake is harmful for patients with PE, since the protein requirement in these patients is relatively higher than in healthy individuals.
Therefore, according to the AASLD/EASL recommendations, daily energy intake should be maintained at 35–40 kcal per 1 kg of body weight, and daily protein intake should be within the range of 1.2–1.5 g/kg [32]. Limiting protein intake is recommended only during the first few days after the development of PE, but then this measure should be abandoned. Animal proteins should be replaced with milk and plant proteins, and foods enriched with branched chain amino acids should be consumed.
Drug therapy
Drug therapy is an important part of the treatment of overt P.E. With minimal PE in the absence of its clinical manifestations, pharmacotherapy, as a rule, is not used. At the same time, it can be prescribed to this category of patients in cases of a noticeable impact of minimal PE on the quality of life. To correct HE, various drugs can be used (non-absorbable disaccharides, antibiotics, branched chain amino acids, L-ornithine-L-aspartate) with varying levels of evidence of effectiveness.
Non-absorbable disaccharides.
Lactulose is a synthetic disaccharide that prevents the formation of ammonia. Lactulose is the drug of first choice in the treatment of overt P.E. Lactulose syrup is prescribed 25 ml every 1-2 hours until at least 2 bowel movements with soft or unformed stools occur during the day. Subsequently, the dose of the drug is titrated individually to maintain 2- or 3-fold bowel movements during the day [35]. Taking excessively high doses of lactulose can lead to complications such as aspiration, dehydration, hypernatremia, irritation of the perianal skin, and in some cases even worsen the course of PE [36].
Antibiotics.
Rifaximin is a non-absorbable antibiotic that inhibits ammoniogenic proteolytic bacterial microflora of the intestine [37]. The results of a number of studies have shown the effectiveness of rifaximin in the treatment of P.E. According to AASLD/EASL guidelines [32], rifaximin is an effective addition to lactulose for the prevention of recurrence of overt PE. In comparative studies, 3-6 months of rifaximin therapy improved cognitive function and reduced ammonemia in patients with PE [38]. Rifaximin is prescribed at a dose of 200-400 mg 2-3 times/day (1200 mg/day) for 5-7 (up to 14) days.
Among other antibiotics, neomycin is still used for the treatment of PE; metronidazole can be used for short-term therapy, however, due to side effects, the long-term use of these drugs is limited [39, 40].
Branched chain amino acids.
A number of studies have shown the effectiveness of therapeutic nutrition with the consumption of food enriched with branched chain amino acids [15]. Due to the preferential absorption of these amino acids, the relative content of aromatic amino acids, which serve as precursors of false neurotransmitters, decreases. In addition, branched chain amino acids help increase muscle mass, resulting in increased ammonia detoxification, which partially occurs in skeletal muscle. The AASLD/EASL guidelines only recommend oral administration of branched-chain amino acids as an alternative or adjunctive treatment for patients who have not responded to conventional therapy [41, 42].
L-ornithine-L-aspartate (LOLA).
Since ornithine and aspartate play a major role in the conversion of ammonia to urea, taking LOLA helps reduce the clinical manifestations of PE. Over the past 10 years, several randomized studies have been conducted, the results of which have shown the high efficacy and safety of this drug in the treatment of PE [43]. LOLA is available as a solution for intravenous infusion and as a granule for oral administration. The standard regimen of use involves intravenous drip administration of 20-30 g of the drug for 7-14 days, followed by transition to oral administration of 9-18 g/day. To achieve a faster and more lasting result, a combination of intravenous and oral administration is possible.
Flumazenil
- an antagonist of benzodiazepine receptors, directly affecting the functions of the central nervous system. Flumazenil is used quite rarely, since giving a short-term effect of restoring consciousness, it promotes only a transient improvement in mental state, but does not affect recovery and survival rates [15].
The effectiveness of the therapy is determined by the reverse development of clinical symptoms.
In case of acute liver failure with the development of hepatic coma and cerebral edema, treatment is carried out in the intensive care unit using the same medications as for chronic liver failure, but in higher doses.
Liver transplantation for cirrhosis with recurrent PE, difficult to respond to conservative therapy, can increase the life expectancy of patients. The average 5-year survival rate of patients after transplantation exceeds 72% [15].
The authors declare no conflict of interest.
Forecast and prevention of the disease
The prognosis for the development of the disease, although it depends on a whole range of factors, is almost always negative. The survival rate of patients is higher if the disease develops against the background of chronic liver pathologies. In cirrhosis, the prognosis is seriously worsened in the presence of jaundice and ascites.
In acute liver failure, the prognosis for the development of the disease worsens in children under 10 years of age and adults after 40 years of age due to viral infection. The mortality rate in the first two stages reaches 30%, and in the last stages it exceeds 80%.
Prevention comes down to normalizing lifestyle, avoiding excessive use of medications and timely treatment of diseases that provoke the development of encephalopathy.
Due to the fact that the lethality of the disease directly depends on the stage of its development, timely detection of the disease is of great importance. To do this, it is important, at the slightest suspicion of the presence of the disease, to seek advice from a specialist, as well as to do a CT and MRI. You can undergo diagnostics at one of the clinics located in the service database. The site has collected information about hundreds of diagnostic clinics. A consultant is ready to help you choose a clinic over the phone and make an appointment for a convenient date.