Published: 06/17/2021 12:50:00 Updated: 06/17/2021
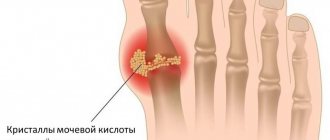
Gout is a systemic disease characterized by impaired purine metabolism in the body and the deposition of urate crystals in the joints. The main manifestation of the disorder is considered to be repeated attacks of arthritis with intense pain in the joints and the appearance of tophi - gouty nodules. Also, with this disease, accumulation of salts in the kidneys is possible with the development of urolithiasis and renal failure.
The prevalence of the disease among adults in Europe ranges from 0.9 to 2.5%, and in the United States reaches 3.9%. Gout is most often diagnosed in men over 40 years of age. Among women, the pathology occurs 6-7 times less often.
Classification
Based on etiology, there are 2 main types of hyperuricemia:
- Primary
- occurs due to a genetically determined defect in enzymes involved in the metabolism of uric acid. - Secondary
– develops against the background of certain diseases, the action of toxins, medications, etc.
According to pathogenesis, there are 3 types of hyperuricemia:
- Metabolic (hyperproductive
) - associated with increased formation of uric acid. - Renal (hypoexcretory
) - due to a slowdown in the release of uric acid from the body in the urine. - Mixed.
According to clinical manifestations there are:
- asymptomatic hyperuricemia;
- acute/intermittent arthritis;
- interictal period;
- chronic tophi gout.
Treatment of gout
The main principle of treating the disease is to reduce hyperuricemia. This is facilitated by changing the patient’s lifestyle and reviewing the diet. Correction of hyperlipidemia, arterial hypertension, hyperglycemia, weight loss and smoking cessation are recommended. A diet for gout involves eliminating foods that are rich in purines or retain them in the body:
- sweet carbonated drinks;
- seafood;
- meat;
- spicy, hot and smoked foods;
- chocolate, ice cream;
- salty cheeses;
- caffeine, cocoa, strong tea;
- alcohol.
Endogenous purines increase when consuming large amounts of animal protein, so the daily requirement for a patient with gout is considered to be no more than 1.5 g per 1 kg of the patient’s body weight. Dairy products with low fat content, such as kefir, cottage cheese, yogurt, help remove uric acid from the body. Polyunsaturated fatty acids, for example, those found in olive oil, have a similar effect. Also, if you have gout, you must follow a drinking regime - at least 2.5 liters of water per day.
It is advisable to begin drug treatment of gout on the first day of the onset of acute arthritis. To stop an attack, non-steroidal anti-inflammatory drugs, colchicine and glucocorticosteroid drugs are used locally or systemically. Hypouricemic therapy is prescribed after laboratory and instrumental confirmation of the diagnosis. It is aimed at:
- reduction of uric acid to the target level - 0.36 mmol/l;
- reduction of foci of urate accumulation in the body;
- reduction in the size of tophi;
- reduction in the frequency of attacks of gouty arthritis.
Allopurinol is considered the drug of choice for this. If it is intolerant, it is possible to use a selective xanthine oxidase inhibitor, febuxostat. Losartan, Amlodipine and Fenofibrate also have a moderate uricosuric effect. Diuretics are used with great caution, only for health reasons. Surgical removal of tophi is carried out only when severe complications occur, for example, carpal tunnel syndrome, spinal compression.
Causes
Physiological reasons
In healthy people, transient or slight increases in uric acid (UA) concentrations may occur without any clinical symptoms. Most often, hyperuricemia is observed when eating meat foods, since meat products are rich in purine bases (adenine, guanine), which are a substrate for the synthesis of uric acid. Hyperuricemia also occurs with dehydration, intense physical activity, and alcohol intake.
Gout
This rheumatological disease can be considered a clinical manifestation of hyperuricemia. Gout develops as a result of a genetically determined defect in enzymes that regulate UA metabolism. Such defects include deficiency of hypoxanthine-guanine phosphoribosyltransferase and increased activity of 5-phosphoribosyl-1-synthetase. Violation of the functional activity of these enzymes leads to increased synthesis of uric acid.
In peripheral tissues, especially in joint capsules and cartilage, crystallization of uric acid salts occurs and their deposition. As a result, the following symptoms appear: acute, intense pain in the joints (usually in the first metatarsophalangeal joint), swelling of the joint with redness of the skin. A fairly specific symptom of gout is the formation of nodules - tophi, localized mainly on the auricles and the back surface of the elbow joints. Gout can also develop against the background of diseases that affect purine metabolism.
Hyperuricemia
Overproduction of uric acid
With intensive destruction of cells and their nuclei, nucleotides are released, from which a large amount of MK is subsequently formed, which leads to an increase in the level of MK. The presence of clinical symptoms of emerging hyperuricemia is determined by the degree of tissue destruction. Increased catabolism is observed in the following diseases and pathological conditions:
- Myeloproliferative and lymphoproliferative diseases (leukemia, lymphoma).
- Tumor lysis syndrome after chemotherapy courses.
- Congenital and acquired hemolytic anemia.
- Severe generalized psoriasis.
- Storage diseases: Gaucher disease, glycogenosis.
Slow elimination of uric acid
Approximately 2/3 of the total amount of MK is excreted in the urine. Therefore, any violation of the excretory function of the kidneys due to their organic damage can lead to hyperuricemia. The same thing often happens when the effective blood volume decreases, which disrupts the blood supply to the kidneys. The following are the main reasons for the suppression of uric acid clearance:
- Glomerulonephritis.
- Interstitial nephritis.
- Chronic renal failure.
- Massive bleeding.
- Congestive heart failure.
- Lead poisoning (lead nephropathy).
- Taking thiazide diuretics.
Other reasons
- Endocrine disorders: hypoparathyroidism, hypothyroidism.
- Diabetic ketoacidosis.
- Lactic acidosis.
- Preeclampsia.
- Sarcoidosis.
- Taking medications: acetylsalicylic acid, cytotoxic drugs (cyclosporine, sodium mycophenolate), anti-tuberculosis drugs (ethambutol, pyrazinamide), nicotinic acid.
Diagnostics
If hyperuricemia is detected, you must consult a general practitioner to determine the cause of its occurrence. At the appointment, the presence of symptoms characteristic of this condition is clarified. The joints that are bothering the patient are carefully examined for swelling or redness. It is determined whether the patient has any chronic diseases, whether he is registered with a dispensary, and what medications he takes. An additional examination is prescribed, including:
- Blood tests.
During a gout attack with severe symptoms, a general blood test shows an increase in ESR and neutrophilic leukocytosis. In biochemical analysis, in addition to hyperuricemia, inflammatory markers are detected (increased C-reactive protein, ferritin); in patients with renal pathology, increased concentrations of creatinine and uric acid are detected. - General urine analysis.
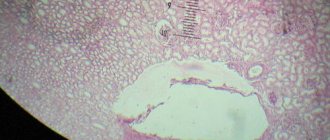
Microscopic examination of urine sediment reveals a large number of urate crystals. In nephrological patients, a chemical analysis of urine may reveal protein, red blood cells, and a change in its relative density. - Study of joint fluid.
To obtain synovial fluid, a puncture of the affected joint is performed. In the native unstained preparation, needle-shaped crystals of monosodium urate are detected by polarization microscopy. This is the gold standard for diagnosing gout. These crystals can also be found in the contents of tophi. - X-ray of joints.
X-ray changes in patients with gout appear only several years after the onset of the disease. Typical radiographic findings include well-defined bone defects in the epiphyses of the feet and hands.
Diet for hyperuricemia
Asymptomatic hyperuricemia – benefit or harm?
Currently, the question is becoming increasingly relevant: how “harmless” is asymptomatic HU [32]? According to some researchers, increasing uric acid can have a beneficial effect on the body. For example, Orowan E. claims that in its chemical structure MK is similar to trimethylated xanthine caffeine and therefore can increase mental and physical performance [33]. Numerous studies in the 1960s and 70s confirmed that people with HU have higher intelligence and reaction speed [34–36]. However, most studies show a relatively small biological effect, and the socioeconomic status of the participants may also have had an influence. Nevertheless, these studies provide some confirmation of the moderate neurostimulating effect of MC. Another useful property of MK is its ability to act as an antioxidant that blocks superoxide, peroxynitrite, and iron-catalyzed oxidative reactions [37]. A number of authors indicate that increased sUA may be one of the key plasma antioxidants and prevent oxidative stress associated with aging, thereby promoting life extension [37]. Studies have shown that infusion of UA in humans increases serum antioxidant activity and improves endothelial function [38]. The ability of GU to reduce peroxynitrite-mediated nitrotyrosine formation indicates the neuroprotective properties of UA, which is especially important in multiple sclerosis, Parkinson's disease, stroke and other neurological diseases. According to epidemiological studies, people with HU are significantly less likely to suffer from multiple sclerosis, Parkinson's disease and Alzheimer's disease, and UA infusion can reduce neurological manifestations obtained experimentally (for example, in experimental allergic encephalomyelitis) [39]. Recent studies have shown that the beneficial effect of UA in these conditions is associated, rather, not with its nitrotyrosine-suppressing antioxidant effect, but with the ability to block the blood-brain barrier or with its effect on astroglial cells [40,41]. Along with data on some positive effects of MK, most studies link HU with the development of cardiovascular diseases (CVD) and kidney damage [42]. Thus, increased sUA is associated with arterial hypertension, increased sodium reabsorption in the proximal renal tubules, microalbuminuria, proteinuria, kidney damage, obesity, hypertriglyceridemia, low high-density lipoprotein cholesterol, hyperinsulinemia, hyperleptinemia, hypoadiponectinemia, damage to the peripheral, carotid and coronary arteries, endothelial dysfunction, oxidative stress, increased concentrations of renin, endothelin and C-reactive protein. SUA levels are higher in postmenopausal women compared to premenopausal women and in urban residents compared to rural residents [31,43,44]. The association between HU and the risk of developing CVD has been established in large epidemiological studies [45–48]. The relationship between HU and arterial hypertension, diabetes mellitus, insulin resistance, obesity and atherosclerosis-related cardiovascular diseases has been proven [45–49]. However, it remains unclear whether HU is a cause or consequence of cardiovascular disease, reflecting the presence of other risk factors such as hypertension, dyslipidemia and diabetes [50]. The NHANES I study (the National Health and Nutrition Examination Survey) revealed an independent association between HU and increased cardiovascular mortality. With an increase in sUA levels, the risk of death from coronary artery disease increased by 77% in men and by 300% in women. An increase in UA concentration of 1 mg/dL (59.5 μmol/L) was associated with a significant increase in mortality from CVD in both men and women [10]. A 12-year study (PIUMA), which included more than 1500 previously untreated patients with hypertension, also demonstrated that serum UA level is a strong predictor of cardiovascular morbidity and mortality [51]. In patients with heart failure, increased sUA levels are considered an indicator of poor prognosis and indicate the need for heart transplantation [52]. A study of patients with type 2 diabetes mellitus showed a significant increase in the incidence of stroke with increasing sUA levels. However, the association remained significant even after excluding other cardiovascular risk factors [53]. Meanwhile, the results of the Framingham study did not reveal a significant association between UA levels and cardiovascular morbidity [50]. The association of sUA levels with target organ damage in patients with hypertension was studied. The results obtained are also ambiguous. A number of studies have found a relationship between GU and left ventricular myocardial mass and other markers of organ damage [54–57]. Other authors did not find a significant association of MB with LV mass, carotid artery disease, or microalbuminuria [58]. In contrast to population data, the results of which regarding the independent significance of uric acid as a risk factor are contradictory; in groups of patients collected according to nosological principles, the significance of GU as a risk factor for the development of CVD and associated complications was established. This applies to patients with gout, as well as patients with rheumatoid arthritis [59] and systemic scleroderma [60]. It has been shown that not only monosodium urate crystals, but also soluble uric acid in asymptomatic HU can lead to an increase in the level of inflammatory mediators and induce the proliferation of vascular smooth muscle cells in vitro [43,61–65]. Many of these “proinflammatory” mediators are of fundamental importance in the pathogenesis of atherosclerotic vascular disease and its complications [64,65]. Currently, several mechanisms have been proposed through which MK can participate in the development of CVD. Thus, Johnson RJ et al. in a series of animal experiments showed that a moderate increase in sUA can cause subtle glomerulotubular damage, promoting activation of the renin-angiotensin system (RAS) and an increase in blood pressure, while all changes reversed after elimination of GU [43, 66]. Sanchez-Lozada LG et al., studying afferent arterioles in rat models of gout, found that high levels of sUA can induce vascular damage, which stopped with the use of allopurinol [67]. It has also been shown that HU causes constriction of renal vessels, correlates with RAS activity [67–69], and is involved in the development of endothelial dysfunction [70]. There is evidence that MK induces proliferation of vascular smooth muscle cells in vitro [62], activating transcription factors and signaling molecules, causing overexpression of cyclooxygenase, platelet-derived growth factor and monocyte chemoattractant protein. UA and the enzyme xanthine oxidase are often detected in atherosclerotic plaques [71,72]. Free radicals encountered in HU stimulate lipid peroxidation, which is responsible for thickening of the carotid intima-media complex [62,68]. Other potential mechanisms by which HU and/or increased xanthine oxidase activity may contribute to vascular injury include platelet adhesion, vascular smooth muscle cell proliferation, and stimulation of the inflammatory response. Thus, HU can exert its pathological effect on blood vessels in various ways [42]. Rats do not develop HU due to the presence of the enzyme uricase, which converts UA into allantoin. Interestingly, suppression of uricase in rats leads to hypertension, mediated by endothelial dysfunction and activation of the renin-angiotensin system, which over time becomes nephrogenic due to the development of arteriolosclerotic vascular damage [73,74]. This is consistent with data from other studies in which HU preceded the onset of hypertension [75,76]. HU was found in almost 90% of adolescents with newly diagnosed hypertension [77]. In pilot studies, a decrease in sUA levels with allopurinol was accompanied by a decrease in blood pressure [78]. Research results indicate that correction of GU can prevent the development of cardiovascular accidents. The LIFE study was the first to show that in patients with hypertension and left ventricular hypertrophy, drug reduction of MBF can reduce cardiovascular risk [79,80]. Thus, the significance of GI in the development of KVZ raises many questions. Thus, if an acute increase in UA has an antioxidant effect and has a beneficial effect on endothelial function, then chronic HU, on the contrary, contributes to the development of oxidative stress and endothelial dysfunction. This may be due to the different intracellular and intravascular effects of acute and chronic HU [81, 82]. Clinical studies have shown that the xanthine oxidase blocker allopurinol can improve endothelial function, reduce cardiovascular complications in patients undergoing coronary artery bypass grafting, and improve cardiac function in patients with dilated cardiomyopathy and congestive heart failure [83–86]. However, other urate-lowering agents have not demonstrated similar results [87,88]. It cannot be ruled out that these effects are due to inhibition of xanthine oxidase and are not associated with a decrease in sUA or that xanthine oxidase inhibitors predominantly affect intracellular sUA, which is associated with most cardiorenal effects [89]. If HU does influence clinical outcomes, then dietary and pharmacological interventions aimed at reducing uric acid may provide a new method for the prevention of cardiovascular disease. Let's hope that further research will soon provide answers to these questions. References 1. Rheumatology: national guidelines, 2008. 2. W Zhang, M Doherty, E Pascual, T Bardin, V Barskova, P Conaghan, J Gerster, J Jacobs, B Leeb, F Liote?, G McCarthy, P Netter, G Nuki, F Perez–Ruiz, A Pignone, J Pimenta?o, L Punzi, E Roddy, T Uhlig, I Zimmermann–Go`rska. EULAR evidence based recommendations for gout. Part I: Diagnosis. Report of a task force of the standing committee for international clinical studies including therapeutics (ESCISIT). Ann Rheum Dis 2006; 65:1301–1311. 3. Campion EW; Glynn RJ; DeLabry LO Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med. 1987; 82(3):421–6. 4. Choi HK, Atkinson K., Karlson EW, Curhan G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the Health Professionals Follow-up Study // Arch Intern Med. – 2005. – No. 165.– rub. 742–748. 5. Dessein PH, Shipton EA, Stanwix AE, Joffe BI, Ramokgadi J. Beneficial effects of weight loss associated with moderate calorie/carbohydrate restriction, and increased proportional intake of protein and unsaturated fat on serum urate and lipoprotein levels in gout: a pilot study // Ann Rheum Dis. – 2000. – No. 59. – p. 539–543. 6. Fam AG Gout, diet, and the insulin resistance syndrome // J Rheumatol. – 2002. – No. 29. – p. 1350–5. 7. Lin KC, Lin HY, Chou P. Community based epidemiological study on hyperuricemia and gout in Kin–Hu, Kinmen // J Rheum.– 2000.– No. 27.– p. 1045–50. 8. Alper AB Jr, Chen W., Yau L., Srinivasan SR, Berenson GS, Hamm LL Childhood uric acid predicts adult blood pressure: the Bogalusa Heart Study // Hypertension. – 2005.– No. 45.– r. 34–38. 9. Grodzicki T., Palmer A., Bulpitt CJ, Incidence of diabetes and gout in hypertensive patients during 8 years of follow-up // J Hum Hypertens. – 1997. – No. 11. – p. 583–585. 10. Fang J., Alderman M. Serum uric acid and cardiovascular mortality: The NHANES I epidemiologic follow-up study, 1971–1992. National Health and Nutrition Examination Survey // JAMA. – 2000. – No. 238. – p. 2404–10. 11. Johnson RJ, Kang DH, Feig D, et al. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? // Hypertension. – 2003. – No. 41. – p. 1183–90. 12. Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal–independent mechanism // Hypertension.– 2001.– No. 38.– p. 1101–6. 13. Tykarski A. Evaluation of renal handling of uric acid in essential hypertension: hyperuricemia related to decreased urate secretion // Nephron.– 1991.– No. 59.– p. 364–368. 14. Breckenridge A. Hypertension and hyperuricaemia // Lancet. – 1966. – No. 1. p. 15–18. 15. Aronoff A. Acute gouty arthritis precipitated by chlorotiazide // N Engl J Med. – 1960. – No. 262. – p. 767–769. 16. Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study // Am J Med. – 1987. – No. 82. – p. 421–426. 17. Grodzicki T., Palmer A., Bulpitt CJ, Incidence of diabetes and gout in hypertensive patients during 8 years of follow-up // J Hum Hypertens. – 1997. – No. 11. – p. 583–585. 18. Lane P. Drug-induced gout // Br. Med J. – 1960. – No. 2. – r. 1383–4. 19. Laragh JH, Heinemann HO Demartini FF Effect of chlorothiazide on electrolyte transport in man; its use in the treatment of edema of congestive heart failure, nephrosis, and cirrhosis // JAMA.– 1958.– No. 166.– p. 145–152. 20. Lin KC, Lin HY, Chou P. Community based epidemiological study on hyperuricemia and gout in Kin–Hu, Kinmen // J Rheum.– 2000.– No. 27.– p. 1045–50. 21. Oren BG, Rich M., Belle MS Chlorothiazide (Diuril) as a hyperuricacidemic agent // JAMA.– 1958.– No. 168.– p. 2128–9. 22. Drum DE, Goldman PA, Jankowski CB Elevation of serum uric acid as a clue to alcohol abuse // Arch Intern Med. – 1981. – No. 141. – p. 477–479. 23. Sharpe CR A case–control study of alcohol consumption and drinking behavior on patients with acute gout // Can Med Assoc J.– 1984.– No. 131.– p. 563–567. 24. Arromdee E, Michet CJ, Crowson CS et al. Epidemiology of Gout: Is the Incidence Rising? //J Rheumatol.– 2002.– No. 29.– p. 2403–6. 25. Adams PF, Hendershot GE, Marano MA Current estimates from the National Health Interview Survey, 1996. – Vital Health Stat 10. – 1999. – 200 rub. 26. Fam AG Gout in the elderly: clinical presentation and treatment // Drug Aging. – 1998. – No. 13. – p. 229–243. 27. Troen BR The biology of aging // Mt Sinai J Med. – 2003. – No. 70. – p. 3–22. 28. US Renal Data System. USRDS 1999 annual data report. Bethesda (MD): National Institute of Health (US), National Institute of diabetes and Digestive and Kidney Diseases.– 1999. 29. Ahn KJ, Kim YS, Lee HC, Park K., Huh KB Cyclosporine–induced hyperuricemia after renal transplant : clinical characteristics and mechanisms // Transplant Proc. – 1992. – No. 24. – p. 1391–2. 30. Saag KG, Choi H. Epidemiology, risk factors, and lifestyle modifications for gout. Arthritis Res Ther. 2006; 8 (suppl 1): S2. 31. Johnson RJ, Titte S, Cade JR, Rideout BA, Oliver WJ. Uric acid, evolution and primitive cultures. Semin Nephrol 2005;25:3–8. 32. Neogi T. Asymptomatic Hyperuricemia: Perhaps Not So Benign? The J of Rheumatol. 2008, 5, 734–737. 33. Orowan E. The origin of man. Nature 1955;175:683–4. 34. Bloch S, Brackenridge CJ. Psychological, performance and biochemical factors in medical students under examination stress. J Psychosom Res 1972;16:25–33. 35. Brooks GW, Mueller E. Serum urate concentrations among university professors; relation to drive, achievement, and leadership. JAMA 1966;195:415–8. 36. Stetten D Jr, Hearon JZ. Intellectual level measured by army classification battery and serum uric acid concentration. Science 1959;129:1737. 37. Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant– and radical–caused aging and cancer: a hypothesis. Proc Natl Acad Sci USA 1981;78:6858–62. 38. Waring WS, McKnight JA, Webb DJ, Maxwell SR. Uric acid restores endothelial function in patients with type 1 diabetes and regular smokers. Diabetes 2006;55:3127–32. 39. Kutzing MK, Firestein BL. Altered uric acid levels and disease states. J Pharmacol Exp Ther 2008;324:1–7. 40. Du Y, Chen CP, Tseng CY, Eisenberg Y, Firestein BL. Astroglia–mediated effects of uric acid to protect spinal cord neurons from glutamate toxicity. Glia 2007;55:463–72. 41. Spitsin SV, Scott GS, Mikheeva T, et al. Comparison of uric acid and ascorbic acid in protection against EAE. Free Radic Biol Med 2002;33:1363–71. 42. Baker JF, Krishnan E, Chen L, Schumacher HR. Serum uric acid and cardiovascular disease: recent developments, and where do they leave us? Am J Med 2005;118:816–26. 43. Johnson RJ, Kang DH, Feig D, et al. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 2003;41:1183–90. 44. Nakanishi N, Okamoto M, Yoshida H, Matsuo Y, Suzuki K, Tatara K. Serum uric acid and risk for development of hypertension and impaired fasting glucose or Type II diabetes in Japanese male office workers. Eur J Epidemiol 2003;18:523–30. 45. Fang J., Alderman M. Serum uric acid and cardiovascular mortality: The NHANES I epidemiologic follow-up study, 1971–1992. National Health and Nutrition Examination Survey // JAMA. – 2000. – No. 238. – p. 2404–10. 46. Freedman DS, Willamson DF, Gunter EW, Byers T. Relation of serum uric acid to mortality and ischemic heart disease. The NHANES 1 Epidemiologic Follow-up Study // Am. J. Epidemiol.– 1995.– No. 141(7).– p. 637–644. 47. Brand FN, McGee DL, Kannel WB et al. Hyperuricemia as a risk factor of coronary heart disease: The Framingham Study // Am. J. Epidemiol. – 1985. Vol. 121. P.11. 48. Niskanen LK, Laaksonen DE, Nyyssonen K. et al. Uric acid level as a risk factor for cardiovascular and all-cause mortality in middle-aged men: a prospective cohort study // Arch. Intern. Med.–2004.–Vol.164, p. 1546. 49. Barskova V.G., Ilyinykh E.V., Eliseev M.S., Zilov A.V., Nasonov E.L. Cardiovascular risk in patients with gout. Obesity and metabolism. 2006, 3(8), 40–44. 50. Culleton BF, Larson MG, Kannel WB, D. Levy. Serum Uric Acid and Risk for Cardiovascular Disease and Death: The Framingham Heart Study // Ann. Intern. Med.– 1999.– No. 131.– r. 7–13. 51. Verdecchia P., Schillaci G., Reboldi G. et al. Relation between serum uric acid and risk of cardiovascular disease in essential hypertension. The PUIMA study. Hypertension 2000; 36:1072–8. 52. Anker SD, Doehner W, Rauchhaus M et al. Uric acid and survival in chronic heart failure: validation and application in metabolic, functional, and hemodynamic staging. Circulation 2003; 107:1991–7. 53. Lehto S., Niskanen L., Ronnemaa T. et al. Serum uric acid is a strong predictor of stroke in patients with non–insulin–independent diabetes mellitus. Stroke 1998; 29: 635–9. 54. Alderman MN, Cohen H, Madhvan S et al. Serum uric acid and cardiovascular events in successfully treated hypertensive patients. Hypertension. 1999; 34: 144–150. 55. Campo C., Ruilope LM, Segura J. Et al. Hyperuricemia, low urine urate excretion and target organ damage in arterial hypertension. Blood press. 2003; 12: 277–283. 56. Viazzi F., Parodi D., Leoncini G. Et al. Serum uric acid and target organ damage in primary hypertension. Hypertension. 2005; 45:991–996. 57. Iribarren C, Folsom AR, Eckfeldt JH et al. Correlates of uric acid and its association with asymptomatic carotid atherosclerosis: the ARIC study. Ann Epidemiol. 1996; 6:331–340. 58. Cuspidi C., Valerio C., Sala C. Et al. Lack of association between serum uric acid and organ damage in a never–treated essential hypertensive population at low prevalence of hyperuricemia. Am J Hypertens. 2007; 20: 678–685. 59. Panoulas VF, Milionis HJ, Douglas KM et al. Association of serum uric acid with cardiovascular disease in rheumatoid arthritis. Rheumatology (Oxford). – 2007; 46(9):1466–1470. 60. Chen CH, Chen HA, Wang HP et al. Pulmonary arterial hypertension in autoimmune diseases: an analysis of 19 cases from a medical center in northern Taiwan. J Micribiol Immunol Infect.–2006; 39 (2): p. 162–168. 61. Leyva F., Anker S., Godsland IF et al. Uric acid in chronic heart failure: a marker of chronic inflammation // Eur. Heart. J. – 1998. Vol.19.– P.1814 –1822. 62. Rao GN, Corson MA, Berk BC Uric acid stimulates vascular smooth muscle cell proliferation by increasing platelet-derived growth factor A-chain expression // J.Biol.Chem.-1991.-Vol. 266.–P.8604–8608. 63. Schumacher HR Jr. Crystal-induced arthritis: an overview // Am J Med. – 1996. – No. 100. – p. 46S–52S. 64. Nasonov E.L. The problem of atherothrombosis in rheumatology. Bulletin of the Russian Academy of Medical Sciences, 2003; 7:6–10. 65. Nasonov E.L., Nasonova V.A., Barskova V.G. Mechanisms of development of gouty inflammation. Ter arch. 2006, 6 (78), 77–84. 66. Johnson RJ, Rodriguez–Iturbe B, Kang DH et al. A unifying pathway for essential hypertension. Am J Hypertens. 2005; 18: 431–440. 67. Sanchez-Lozada LG, Tapia E, Avila-Casado C. Et al. Mild hyperuricemia induces glomerular hypertension in normal rats. Am J Physiol. 2002; 283:F1105–F1110. 68. Mazzali M., Kanellis J., Han L. Et al. Hyperuricemia induces a primary arteriolopathy in rats by a blood pressure–independent mechanism. Am J Physiol. 2002; 282:F 991–F997. 69. Saito I., Saruta T., Kondon K. Et al. Serum uric acid and the renin–angiotensin system in hypertension. J Am Geriatr Soc. 1978; 26:241–247. 70. Waring WS, Webb DJ, Maxwell SRJ et al. Effect of local hyperucemia on endothelial function in the human forearm vascular bed. Br J Clin Pharmacol. 2000; 49:511. 71. Suarna C., Dean RT, May J. et al. Human atherosclerotic plaque contains both oxidized lipids and relatively large amounts of alpha–tocopherol and ascorbate. Arterioscler Thromb Vasc Biol. 1995; 15: 1616–1624. 72. Iribarren C, Folsom AR, Eckfeldt JH et al. Correlates of uric acid and its association with asymptomatic carotid atherosclerosis: the ARIC Study: Atherosclerosis Risk in Communities. Stroke. 1999; 29:635–639. 73. Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal–independent mechanism. Hypertension 2001;38:1101–6. 74. Watanabe S, Kang DH, Feng L, et al. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension 2002;40:355–60. 75. Masuo K, Kawaguchi H, Mikami H, Ogihara T, Tuck ML. Serum uric acid and plasma norepinephrine concentrations predict subsequent weight gain and blood pressure elevation. Hypertension 2003;42:474–80. 76. Sundstrom J, Sullivan L, D'Agostino RB, Levy D, Kannel WB, Vasan RS. Relations of serum uric acid to longitudinal blood pressure tracking and hypertension incidence. Hypertension 2005;45:28–33. 77. Feig DI, Johnson RJ. Hyperuricemia in childhood primary hypertension. Hypertension 2003;42:247–52. 78. Feig DI, Nakagawa T, Karumanchi SA, et al. Hypothesis: Uric acid, nephron number, and the pathogenesis of essential hypertension. Kidney Int 2004;66:281–7. 79. Dahlof B., Deveereux Rb, Kjeldsen Se et al. Cardiovascular Morbidity and Mortality in the Losartan Intervents for Endpoint Reduction in Hypertension Study (Life): A Randomized Trial AGAINST Atenolol. Lancet 2002; 359: 995–1003. 80. Hoieggen A., Alderman MH, Kjeldsen Se et al. The Impact of Serum Uric Acid on Cardiovascular Outcomes in the Life Study. Kidney int. 2004; 65: 1041–1049. 81. Santos CX, Anjos EI, Augusto O. Uric Acid Oxidation by Peroxynite: Multiple Reactions, Free Radical ForMation, and Amplification of Lipid Oxidation. Arch Biochem Biophys 1999; 372: 285–94. 82. Sautin Yy, Nakagawa T, Zharikov S, Johnson Rj. Adverse Effects of the Classical Antioxidant Uric Acid in Adipocytes: Nadph Oxidase - Medicate/Nitrosative Stress. Am J Physiol Cell Physiol 2007; 293: C584–96. 83. Doehner V., Schoene N., Rauchhaus M. et al. Effects of Xantine Oxidase Inhibition with Allopurinol On Endothelial Function and Peripheral Blood Flow in Hyperuricemic Paty Chronic Heart Failure: Results From 2 Plath From 2 PLACO 2 PLEC -Controlled studies. Circulation 2002; 105: 2619–2624. 84. Coghlan JG, Flitter WD, Clutton SM et al. Allopurinol Pretreatment Imprings Postoperation Recovery and Reduces Lipid Peroxidation in Patiegoint Coronary Artry Bypass Grafting. J Thorac Cardiovasc Surg. 1994; 107: 248–256. 85. Weimert Na, Tanke Wf, Sims JJ. Allopurinol as a Cardioprotective During Coronary Artery Bypass Graft Surgery. Ann Pharmacother 2003; 37: 1708–1711. 86. Cappola tp, kass da, nelson gs et al. Allopurinol IMPROPROVES MyCardial Effecency in Patents with Idiopathic Dilated Cardiomyopathy. Circulation 2001; 104: 2407–2411. 87. George J, Carr E, Davies J, Belch JJ, Struthers A. High - Dose Allopurinol Improvs Endothelial Function by Profundly Reducular Oxidate Stress and Not By Lowering Uring IC Acid. Circulation 2006; 114: 2508–16. 88. Waring WS, McKnight Ja, Webb DJ, Maxwell SR. Lowering Serum Urate Does Not Improve Endothelial Function in Patience with Type 2 Diabetes. Diabetologia 2007; 50: 2572–9. 89. Kang DH, Park SK, Lee IK, Johnson Rj. Uric Acid - Induced C - Reactive Protein Expression: Implication On Cell Proliferation and Nitric Oxide OF HUMAN VASCULAR CELLS. J Am Soc Nephrol 2005; 16: 3553–62.
Correction
Drug treatment
To correct hyperuricemia, it is necessary to eliminate the etiological factor. If, during the collection of anamnesis, it turns out that the patient is taking a drug that causes hyperuricemia, one should consider either reducing the dosage or replacing the drug with an alternative one that does not have such side effects. Treatment tactics are largely determined by the presence of clinical symptoms, as well as their severity.
- Diet.
People suffering from gout should definitely stop drinking alcohol and exclude high-purine foods (red meat, liver, smoked meats) from their diet, as they can provoke an increase in symptoms. - Colchicine alkaloids.
The main group of drugs for the relief of acute gout attacks. Reduction of symptoms occurs within 12 hours after the first dose. - NSAIDs.
Prescribed in addition to colchicine alkaloids to relieve arthritis symptoms. Preference is given to drugs with the most pronounced analgesic and anti-inflammatory effect. - Glucocorticosteroids.
Hormonal drugs are used to eliminate symptoms in case of individual intolerance to colchicine and NSAIDs or if the patient has strict contraindications to their use. - Uricodepressors.
Drugs in this group reduce the content of UA by suppressing the activity of the enzyme involved in the synthesis of UA (xanthine oxidase). Long-term use helps reduce the incidence of gout symptoms, slow down the deposition of crystals and the formation of tophi. - Selective xanthine oxidase inhibitors.
The main difference from uricodepressors is the lower incidence of unwanted side reactions, such as upper and lower respiratory tract infections, hepatotoxicity, and hypersensitivity reactions. - Uricosuric drugs.
These drugs inhibit the reabsorption of sUA in the renal tubules, thereby increasing the excretion of sUA. - Anti-gout drugs of mixed action.
There are also combination drugs that simultaneously act on 2 levels of urinary magnesium metabolism. They contain a uricodepressive component and a uricosuric agent. - Alkalizing drugs.
To prevent the development of urate nephropathy and the deposition of urate stones, the main urate-lowering therapy should be supplemented by taking drugs to alkalinize urine - sodium citrate, sodium bicarbonate.
Experimental therapy
Development and clinical trials of new drugs to combat hyperuricemia continue. Rasburicase, a drug containing recombinant uricase (an enzyme that destroys uric acid), has shown high effectiveness in the treatment of tumor lysis syndrome. This drug is also distinguished by the possibility of resorption of deposited sodium monourate crystals.
Promising is the use of genetically engineered biological drugs - an antagonist of interleukin-1 receptors (anakinra), fully humanized monoclonal antibodies to IL-1-beta (canakinumab), soluble protein IL-1 (rilonacept). Their use has demonstrated sufficient effectiveness in preventing exacerbations of symptoms of chronic gouty arthritis.