Pulmonary hypertension - a disease characterized by increased blood pressure pulmonary artery system, which can be caused by a significant increase in the volume of pulmonary blood flow or an increase in resistance in the vascular bed of the lungs.
Pulmonary hypertension is almost always a secondary condition. If doctors have not been able to identify the cause, then pulmonary hypertension in this case is considered to be primary. With this type, the pulmonary vessels narrow, their hypertrophy and fibrization occur. Due to pulmonary hypertension, right ventricular overload and failure develop.
- Causes and epidemiology
- Pathogenesis
- Classification
- Symptoms
- Diagnostics
- Treatment
- Forecast
Symptoms include shortness of breath on exertion, fatigue, possible fainting and chest discomfort. Diagnosis is made by measuring pulmonary artery pressure. Treatment is carried out with vasodilators, and in severe cases they resort to a lung transplant. In general, the prognosis is poor for primary pulmonary hypertension, since therapy cannot be carried out to eliminate the underlying pathology.
Normal pulmonary artery pressure readings :
- systolic 23-26 mm Hg.
- diastolic 7-9 mm Hg.
- average 12-15 mm Hg.
According to WHO recommendations, the maximum normal systolic pressure in the pulmonary artery should be 30 mm Hg, and the maximum diastolic pressure should be 15 mm Hg.
Pathogenesis
Primary PH is characterized by smooth muscle hypertrophy, variable vasoconstriction, and vessel wall remodeling. Vasoconstriction is caused by an increase in the activity of thromboxane and endothelin 1, as well as a decrease in the activity of nitric oxide and prostacyclin. Increased pulmonary vascular pressure is caused by vascular obstruction. It affects endothelial damage.
As a result, coagulation on the intimal surface is activated, which can aggravate arterial hypertension. This is also facilitated by thrombotic coagulopathy, which is a consequence of an increase in the content of plasmogen activator inhibitor type 1 and fibrinopeptide A and a decrease in the activity of tissue plasmogen activator. Focal coagulation on the surface of the endothelium must be distinguished from chronic thromboembolic pulmonary arterial hypertension, which is provoked by organized pulmonary thromboembolism.
As a result, in most patients, primary pulmonary hypertension becomes a provoking factor of right ventricular hypertrophy with dilatation and right ventricular failure.
Risk factors
As you get older, your risk of developing chronic pulmonary hypertension increases. The disease is more often diagnosed in people aged 30 to 60 years. However, the idiopathic form is more common in young people.
Other factors that may increase the risk of pulmonary hypertension include:
- family medical history;
- excess weight;
- bleeding disorders or a family history of blood clots in the lungs;
- exposure to asbestos;
- genetic disorders, including congenital heart defects;
- life at high altitude;
- using certain drugs for weight loss;
- use of illegal and narcotic drugs;
- use of selective serotonin reuptake inhibitors, used to treat depression and anxiety.
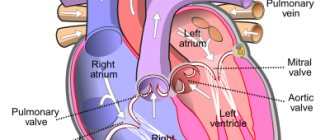
Classification
Left ventricular failure
- Arterial hypertension
- Myocarditis
- Cardiomyopathies
- Mitral regurgitation
- Coarctation of the aorta, aortic valve defects
- Cardiac ischemia
Increased pressure in the left atrium
- Tumor or thrombosis of the left atrium
- Mitral stenosis
- Supravalvular mitral annulus, triatrial heart
Pulmonary vein obstruction
- Pulmonary venous thrombosis
- Mediastinal fibrosis
Parenchymal lung diseases
- Interstitial lung diseases
- Chronic obstructive pulmonary diseases (COPD)
- Acute severe lung injury (severe diffuse pneumonitis, adult respiratory distress syndrome)
Diseases of the pulmonary artery system
- Repeated or massive pulmonary embolisms
- Primary pulmonary hypertension
- Systemic vasculitis
- Thrombosis "in situ" of the pulmonary artery
- Increased pulmonary blood flow volume (patent ductus arteriosus, congenital heart disease with left-to-right shunting)
- Distal pulmonary stenosis
- Pulmonary hypertension caused by food or drugs
Pulmonary hypertension of newborns
- Hyaline membrane disease
- Persistent fetal circulation
- Meconium aspiration
- Diaphragmatic hernia
Hypoxia and/or hypercapnia
- Obstruction of the upper respiratory tract (obstructive sleep apnea syndrome, enlarged tonsils)
- Accommodation high in the mountains
- Primary alveolar hypoventilation
- Pickwick's syndrome (hypoventilation in fat people)
Many authors classify such forms of pulmonary hypertension as acute and chronic based on the timing of development.
Causes
The main diseases that provoke the development of pulmonary hypertension and cor pulmonale syndrome are chronic lung diseases. Most often these are bronchopulmonary diseases:
- chronic obstructive bronchitis (characterized by changes in lung tissue);
- bronchiectasis (distinguished by the formation of cavities in the lower parts of the lungs);
- fibrosis of lung tissue, which is caused by pathological processes in the parenchyma, etc.
In addition, heart disease may be a cause:
- certain birth defects;
- diseases that impair the functionality of the heart muscle.
Also, periodic thrombosis of the pulmonary artery vessels is also often the cause of the development of the syndrome. There are several main pathways in the development of the disease:
- alveolar hypoxia (lack of oxygen);
- changes in the structure of lung tissue;
- increase in the number of red blood cells.
There are other factors that can cause pulmonary hypertension:
- certain medications, toxins;
- medical and demographic factors (gender, condition, presence of diseases).
The development of pulmonary hypertension can be triggered by the presence of the following diseases:
- HIV infection;
- cirrhosis of the liver;
- blood diseases;
- portal hypertension;
- hyperthyroidism;
- some hereditary diseases;
- compression of the blood vessels of the lungs (by a tumor, a changed chest or as a result of obesity);
- physical activity, especially lifting in height.
Symptoms
The first clinical manifestations occur when there is a more than twofold increase in blood pressure in the pulmonary artery. The main symptoms are almost identical for pulmonary hypertension of any etiology. Main complaints:
- increased fatigue and weakness in the body
- shortness of breath (appears at the very beginning of the disease), first during physical activity, later at rest
- persistent pain in the heart area (caused by relative coronary insufficiency)
- fainting (due to a lack of oxygen in the human brain, typical for the primary form of the disease in question)
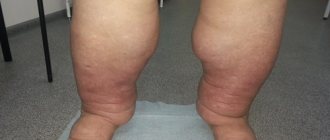
- pain in the liver and swelling in the feet and legs
- Hoarseness of voice (rare)
- hemoptysis (often, especially with a significant increase in pressure in the pulmonary artery)
The vast majority of cases are characterized by rapid fatigue and progressive shortness of breath during exertion. With shortness of breath, there may be atypical discomfort in the chest, and during physical activity, dizziness and even faintness are common. Such manifestations are mainly explained by insufficient cardiac output. Raynaud's phenomenon occurs in approximately 10 cases out of 100 with primary pulmonary hypertension, and 99% of such cases occur in women. A symptom such as hemoptysis is rarely noted; it most often signals a future death. Rarely, dysphonia occurs due to compression of the recurrent laryngeal nerve by the enlarged pulmonary artery (Ortner's syndrome).
In advanced cases, pulmonary hypertension can manifest itself with the following symptoms:
- diffuse second tone (S2) with an emphasized pulmonary component S (P)
- right ventricular bulge
- third sound of the right ventricle (S3)
- pulmonary ejection click
- swelling of the jugular veins
- peripheral edema
- congestion in the liver
Portopulmonary hypertension
This is severe pulmonary arterial hypertension with portal hypertension in patients without secondary causes. Pulmonary hypertension is observed in patients with various conditions that lead to portal hypertension without or with cirrhosis. Portopulmonary hypertension is less common compared to hepatopulmonary syndrome in people with chronic liver diseases; according to statistics, this is from 3.5 to 12% of cases.
The first signs to appear are increased fatigue and shortness of breath. ECG and physical examination methods reveal manifestations characteristic of PH. Regurgitation is often recorded at the tricuspid valve. This diagnosis is suspected upon receipt of EchoCG data, confirmation is carried out using catheterization of the right heart. Treatment requires elimination of primary pulmonary hypertension without prescribing hepatotoxic drugs. In some cases, the doctor resorts to vasodilator therapy. The outcome depends on the underlying liver disease.
Portopulmonary hypertension is a relatively contraindication to liver transplantation, since there is a high risk of complications and death for the patient. After transplantation, in some cases with moderate pulmonary hypertension, the disease reverses.
Interview and examination of the patient
It is necessary to find out whether the patient has had episodes of deep vein thrombosis, sudden swelling of the legs, or thrombophlebitis. Many patients have a family history of sudden death, cardiovascular disease, and an increased tendency to thrombus formation.
Objective evidence of a history of pulmonary embolism (PE) is the coincidence in time of the clinical presentation of thrombosis of the veins of the lower extremities and the appearance of shortness of breath. In the coming months after PE, a period can be identified in patients when the condition remains stable and asymptomatic. This is due to the fact that the right ventricle copes with the load and allows maintaining good exercise tolerance until progressive pulmonary vascular remodeling develops. Almost the only reliable evidence of previous PE can be data from perfusion scintigraphy or computed tomography of the lungs performed during an acute episode of PE.
During a general examination of patients with CTEPH, blueness (cyanosis) may be detected. With the development of right ventricular heart failure, swollen neck veins, enlarged liver, peripheral edema, and abdominal dropsy are noted.
Diagnostics
Objective examination reveals cyanosis. Developing for a long time without treatment, the disease leads to the fact that the distal phalanges of the fingers take on the appearance of “drumsticks”, and the nails take on the shape of “watch glasses”. Auscultation of the heart reveals typical signs: accent (maybe splitting) of the second tone over a.pulmonalis; systolic murmur over the area of the xiphoid process, increasing with inspiration. At the peak of the development of the disease, there may be a diastolic murmur in the second intercostal space on the left, which is explained by the relative insufficiency of the pulmonary valve with its strong expansion.
Cardiac percussion does not reveal signs typical of pulmonary hypertension, and therefore is not considered an effective diagnostic method in these cases. Rarely, an expansion of the border of vascular dullness in the second intercostal space on the left and a displacement of the right border of the heart outward from the right parasternal line are recorded (which is explained by hypertrophy of the right ventricular myocardium). With pulmonary hypertension, hypertrophy of the pancreas and right atrium is usually found, as well as signs that indicate an increase in pressure in the pulmonary artery.
To detect these signs, resort to the following methods:
- ECG,
- X-ray of chest organs,
- catheterization of the right heart with measurement of pressure in the right atrium, right ventricle, and in the trunk of the pulmonary artery
- EchoCG
To detect the causes of the disease, other methods are used :
- CT lungs
- X-ray tomography of the lungs
- angiopulmonography
- ventilation-perfusion radionuclide scintigraphy of the lungs
The above methods help to detect pathology of the parenchyma and vascular system of the lungs. In some cases, diagnosis is also carried out using a lung biopsy to detect pulmonary veno-occlusive disease, diffuse interstitial lung diseases, pulmonary capillary granulomatosis, etc.
Among the symptoms of PH, there may be hypertensive crises in the pulmonary artery system , which are manifested by the following symptoms:
- severe cough, sputum may contain blood
- severe suffocation (mainly in the evening/night hours)
- pronounced general cyanosis
- orthopnea (severe shortness of breath associated with stagnation in the pulmonary circulation)
- fast and weak pulse
- excitement (not always)
- bulging of the conus a.pulmonalis
- pronounced pulsation of a.pulmonalis in the 2nd intercostal space
- accent of the second tone on a.pulmonalis
- pulsation of the right ventricle in the epigastrium
- the appearance of vegetative reactions in the form of urina spastica, involuntary defecation after the end of the crisis
- swelling and pulsation of the neck veins
- appearance of the Plesch reflex
Primary pulmonary hypertension is suspected if the patient has severe dyspnea on exertion and there is no history of other diseases that could lead to PH. First, a chest X-ray, ECG, and spirometry are done to look for the most common causes of shortness of breath. After this, Doppler echocardiography is used to measure pressure in the right ventricle and pulmonary arteries and detect possible anatomical abnormalities that may be the cause of secondary PH.
In primary pulmonary hypertension, radiographs often reveal expansion of the roots of the lungs with a pronounced narrowing towards the periphery. Spirometric indicators and lung volumes may be normal or moderately limited. But the diffusivity of carbon monoxide (DL) is usually below normal. The ECG shows such general changes as include deviation of the electrical axis to the right, R > S in V; SQ T and peak P waves.
To detect secondary causes that cannot be determined by symptoms, the doctor prescribes additional tests. This includes perfusion-ventilation scanning to detect thromboembolic disease; pulmonary function tests (makes it possible to detect restrictive or obstructive pulmonary diseases); serological tests (to rule out or confirm rheumatic disease).
Chronic thromboembolic pulmonary arterial hypertension can be suggested by appropriate findings on computed tomography or pulmonary scans and is diagnosed by arteriography. If necessary, these methods are also used:
- polysomnography
- liver function tests
- HIV test
If the initial examination does not reveal conditions associated with secondary PH, the doctor performs a pulmonary artery catheterization. This measures pressure in the right side of the heart and pulmonary artery, pulmonary capillary wedge pressure, and cardiac output. To exclude atrial septal defect, blood O2 saturation in the right side is measured.
During the procedure, vasodilating drugs are often used (intravenous epoprostenol, adenosine, inhaled nitric oxide, etc.). If the body responds to the administration of the above-mentioned drugs by reducing the pressure in the right side, this indicates that such a drug will be effective in treating the condition. Previously, a biopsy was often used, but today doctors do not advise resorting to this method due to the large number of possible complications and the high percentage of deaths.
If a diagnosis of primary pulmonary hypertension is made, the patient's family history should be examined to detect possible genetic transmission (as indicated by cases of premature death of relatively healthy family members). For familial primary pulmonary hypertension, genetic counseling should be consulted to inform family members of the risk of the disease (approximately 20%). They are recommended to undergo echocardiography for evaluation. Testing for mutations in the BMPR2 gene in familial primary pulmonary hypertension may be of value in the future.
The role of regional centers and LG offices
The main task of regional centers is to determine the PH group (according to the classification), its hemodynamic parameters, and also decide on the prescription of specific therapy.
Patients who apply to these centers should be further examined. PH is most common in the population due to pathology of the left side of the heart and pathology of the lungs. This determines the diagnostic search. All patients should undergo an echocardiogram according to a special protocol, an electrocardiogram (ECG), a chest x-ray, a complete blood count, a biochemical blood test, blood gas determination, NT-proBNP (natriuretic peptide) level, spirometry (if not previously done on an outpatient basis). outpatient department), 6-minute walk test; according to indications - cardiorespiratory exercise test, computed tomography, coronary angiography, etc.
If a patient has a pathology of the left heart or lung disease in the absence of severe right ventricular dysfunction and severe PH, he should be recommended periodic echocardiography monitoring (usually every 6-12 months or if the condition worsens) and treatment of the underlying disease should be started. .
It should be remembered that the patient may have a mixed etiology of PH, therefore, if the severity of the underlying disease and right ventricular dysfunction do not correspond, he should be referred to the Federal Expert Center.
If a patient is suspected of having CTEPH, he needs ventilation-perfusion lung scintigraphy. If such equipment is not available, the possibility of sending it to MSCT angiography (multispiral computed tomography), which is available in most regional centers, should be considered. Also, according to indications, such patients are diagnosed with coagulopathies [1].
If systemic connective tissue diseases are suspected, primarily systemic scleroderma, tests for antinuclear and anticentromere antibodies, as well as antibodies to Scl-70 (topoisomerase I) are necessary [1].
According to the hemodynamic definition, PH can be precapillary, isolated postcapillary and combined pre- and postcapillary (Table 2) [2]. To diagnose it, it is necessary to carry out invasive catheterization of the right heart, which is available in expert or large regional medical centers. In order to evaluate the possibility of prescribing calcium channel blockers to patients during this procedure, a vasoreactive test is also performed [2].
After diagnosing the PH group, all patients need to have their risk of adverse outcomes assessed over the next 12 months. (Table 3). It is recommended to determine the patient's risk at each doctor's visit. It should be noted that this algorithm was developed based on data obtained from studies in patients with idiopathic PAH and has not been validated for other forms of PH [1].
Treatment
Treatment of secondary pulmonary hypertension is aimed at getting rid of the underlying disease. Patients with severe pulmonary arterial hypertension due to chronic thromboembolism are treated with pulmonary thromboendarterectomy. This operation is considered more complex than emergency surgical embolectomy. Under conditions of extrapulmonary circulation, an organized vascularized thrombus is excised along the pulmonary trunk. In specialized centers, the mortality rate during and after surgery is less than 10%.
Treatment of primary pulmonary hypertension should begin with oral calcium channel blockers. In a minority of patients, these drugs reduce pulmonary artery pressure or pulmonary vascular resistance. Most doctors do not recommend verapamil because it has a negative inotropic effect. If treatment with calcium channel blockers is effective, the prognosis is favorable, you need to continue the course. If there is no response to treatment, the doctor prescribes other drugs.
Intravenous epoprostenol (a prostacyclin analogue) is often used to treat pulmonary hypertension. It increases survival in patients, even those who do not respond to vasodilator medications during catheterization. This treatment has disadvantages, such as the need for an indwelling central catheter. Side effects also occur, such as bacteremia and diarrhea. Oral (beraprost), inhaled (iloprost), subcutaneous (treprostinil) analogs of prostacyclin are considered alternative drugs. But their effects on the human body are still being studied, so they are not widely introduced into medical practice.
The oral endothelin receptor antagonist bosentan is effective in some cases. It is mainly prescribed for milder forms of pulmonary hypertension and insensitivity to vasodilators. L-arginine and oral sildenafil are also currently under investigation.
Lung transplantation is a dangerous procedure that can cause complications, because it is possible that an infection may occur and the body may reject the new lung. The procedure is used for heart failure of the fourth degree (according to the classification of the New York Heart Association), when a person develops shortness of breath even with minimal activity, due to which he cannot actively live, but constantly lies or sits; and only if in such cases there is no effectiveness of prostacyclin analogues.
Many patients are prescribed additional medications to treat deficiency, including diuretics. They should also receive warfarin to prevent thromboembolism.
PH therapy
The treatment of patients with PH is based not only on the prescription of specific therapy, but also on a number of other measures that can improve the patient’s prognosis.
It is advisable to individually select the level of physical activity for patients with PH. It should not cause severe shortness of breath, dizziness, syncope, or chest pain [4]. Vaccination against influenza and pneumococcus should be recommended to all patients with PAH. When traveling by air, due to the high risk of developing vasoconstriction due to hypoxia, it is necessary to provide oxygen for the patient. Women with PAH are recommended to terminate pregnancy due to the high risk of adverse outcomes [5].
Also, patients with PH need maintenance therapy aimed at treating heart failure and improving the prognosis.
For anemia of any degree, correction of hemoglobin levels is necessary, since individuals with PH are highly sensitive to its decrease. In case of erythrocytosis with a hematocrit of more than 65% (in the presence of symptoms - headaches, impaired concentration), developed against the background of long-term hypoxia, phlebotomy is possible. In other cases, phlebotomy should be avoided [1].
Long-term oxygen therapy is indicated when oxygen saturation decreases to less than 90%. It is most effective in patients with PH due to lung disease. In people with Eisenmenger syndrome, according to studies, oxygen therapy does not affect hematological parameters, quality of life and survival [6].
In case of decompensation of right ventricular failure, patients are prescribed diuretic therapy in accordance with the recommendations for the management of patients with chronic heart failure (CHF) [7]. Renal function and blood electrolytes should be regularly assessed in order to promptly adjust the dose of diuretics.
Angiotensin-converting enzyme inhibitors (ACEIs) and beta blockers in patients with PAH have been shown to have little effect and are therefore not currently used. In addition, the use of these drugs, even in small doses, can lead to the development of hypotension and worsening of right ventricular failure [8].
Oral anticoagulants (OAC) can be used in some patients with PH. In idiopathic PAH they significantly improve survival (hazard ratio 0.79, 95% confidence interval 0.66–0.94). They are also indicated in patients with CTEPH, inherited PAH and associated PAH while taking anorectics. In other forms of PH, in particular PAH associated with systemic connective tissue diseases, portopulmonary hypertension and PAH associated with congenital heart defects (CHD), the risk of bleeding may be increased, which requires an individual approach to the prescription of this group of drugs [1].
When myocardial contractility is reduced in patients with PAH, cardiac glycosides may be useful. Intravenous administration of digoxin in this group of patients causes a moderate increase in cardiac output. Patients with atrial fibrillation/flutter can, in some cases, take cardiac glycosides to control heart rate. The use of inotropic drugs (eg, dopamine), as in left ventricular failure, may be effective in patients with PH [9].
If it is necessary to prescribe antiarrhythmic drugs, primarily for atrial fibrillation/flutter, preference should be given to drugs without a negative inotropic effect, such as amiodarone [1].
If a patient has PH associated with damage to the left side of the heart or lung diseases, the patient, as a rule, is not indicated for specific therapy [8].
Forecast
Without treatment, a person with pulmonary hypertension lives on average 2.5 years. The cause of death was sudden death due to right ventricular failure. Five-year survival with epoprostenol treatment is observed in more than half of patients. And when treatment with calcium channel blockers is effective, the 5-year survival rate is 90% or more.
Pulmonary hypertension has a poor prognosis if symptoms such as low cardiac output, higher pulmonary artery and right atrium pressures, lack of response to vasodilators, heart failure (HF), hypoxemia, and deterioration in overall functional status are present.
Degrees of pulmonary hypertension
Depending on the stage of development of the disease, its degrees are distinguished.
- Pulmonary hypertension stage 1. There are no clinical manifestations, physical activity is well tolerated, but in some areas of the pulmonary artery there are signs of circulatory disorders that can only be detected with a special examination.
- Pulmonary hypertension 2 degrees. Physical activity begins to bring discomfort. Worried about shortness of breath, weakness, dizziness. At rest I feel normal.
- Pulmonary hypertension grade 3. Even light physical activity causes severe discomfort.
- Pulmonary hypertension grade 4. All symptoms of pulmonary hypertension are felt even at rest; a person cannot lead a normal lifestyle.
Complications of the disease
The most common complication of pulmonary hypertension in both newborns and adults is the development of right ventricular heart failure, which is accompanied by atrial fibrillation. With a more severe course of the disease, thrombosis of the pulmonary arterioles may develop.
Also, complications from pulmonary disease increase the risk of developing a hypertensive crisis, which is accompanied by attacks of suffocation, especially at night, pulmonary edema, severe coughing with an attack of hemoptysis may develop.
In this case, a hypertensive crisis can result in involuntary emptying of the bladder or defecation.
And as a result of arterial thromboembolism, the lung may cease to perform its function, which leads to the death of the patient.
Why is it developing?
Pulmonary arterial hypertension occurs in two types, depending on the cause of its occurrence.
Primary or idiopathic
The etiology of the disease has not yet been precisely established, and the form of the pathology is quite rare. Severe idiopathic hypertension is first diagnosed in newborns, having a hereditary form. Pathology can be transmitted not only in a direct line from the mother or father, but also from grandparents or even great-grandparents.
During diagnosis, children with a similar diagnosis are found to have atherosclerotic changes in the cavity of the pulmonary artery and an increase in the mass of the right cardiac ventricle. If a child or adult who first develops pulmonary hypertension syndrome is not given immediate assistance, he will remain disabled, and complete ignorance of the symptoms can lead to the death of the patient.
Secondary
It develops more often in adults against the background of concomitant diseases. Secondary pulmonary hypertension is observed in pathologies occurring against the background of insufficient functioning of the left ventricle, HIV infection and heart defects of congenital etiology. The risk of developing the disease increases for people with connective tissue pathologies - lupus erythematosus, systemic scleroderma.
With acquired heart defects, after thromboembolism has been transferred to the pulmonary artery cavity, there is also a high risk of signs of increased blood pressure in the vascular bed of the pulmonary arteries. In lung diseases, pathology can also manifest itself, but usually does not reach an advanced stage. Potential dangers are those conditions that increase the risk of developing pulmonary hypertension.
This is taking drugs from certain drug groups (Aminorex, Amphetamines, Cocaine, Fenfluramine, Chemotherapy drugs); oral contraceptives, antidepressants, and estrogen drugs are considered less dangerous in this regard. Diseases that increase the chances of developing pulmonary hypertension are HIV infection, the presence of congenital shunts located between the systemic and pulmonary capillaries, severe diseases of the thyroid gland and liver, genetic mutations and metabolic disorders.
No ads 1
Clinical observation
Patient K., 42 years old, applied for an outpatient appointment at the PH office of a regional center with complaints of shortness of breath, weakness, and periodic chest pain not associated with physical activity. 10 years ago, a congenital heart defect, multiple ventricular septal defects, and a bicuspid aortic valve were discovered. Eisenmenger syndrome. High LH. Moderate aortic valve stenosis. CHF stage 2A, FC 3. Right-sided scoliosis.
Upon examination, the condition is of moderate severity at the moment. The skin is pale, with a cyanotic tint. Auscultation: vesicular breathing in the lungs, no wheezing, respiratory rate = 20 per minute. Heart: clear tones, regular rhythm, systolic murmur in the projection of the aortic valve. BP=110/70 mm Hg. art., heart rate = 78 beats/min. The abdomen is soft and painless. The liver is +3.0 cm from under the costal arch, the edge is dense, the spleen is not enlarged. Oxygen saturation - 84–87%. 6-minute walk test - 174 m. The patient is constantly taking torasemide 5 mg/day, eplerenone 25 mg/day, diltiazem 90 mg/day, warfarin 2.5 mg/day under the control of an INR of 2.0–3.0.
General blood test: hemoglobin - 161 g/l, erythrocytes - 7.6 × 1012/l, hematocrit - 56%, average erythrocyte volume - 73.6 fl, average hemoglobin content in an erythrocyte - 21.1 pg, average hemoglobin concentration in erythrocytes - 287 g/l, leukocytes - 4.8x109/l, platelets - 160x109/l.
Biochemical blood test: total protein - 75 g/l, creatinine - 58 µmol/l, uric acid - 394.8 µmol/l, aspartate aminotransferase (AST) - 16.1 U/l, alanine aminotransferase (ALT) - 15.6 U /l, total bilirubin - 10.5 µmol/l, potassium - 4.3 mmol/l.
EchoCG: no violations of local contractility. Left ventricular ejection fraction - 65%. Dilatation of the left and right atrium, right ventricle, multiple ventricular septal defects, bicuspid aortic valve. Eisenmenger syndrome. High pulmonary hypertension (average pulmonary artery pressure - 88 mm Hg). Moderate aortic valve stenosis.
Taking into account the lack of compensation and the ineffectiveness of the therapy, the patient was prescribed Bozenex 62.5 mg twice a day under the control of AST, ALT, and bilirubin. According to the recommendations, bosentan can be recommended for patients with Eisenmenger syndrome FC 3 according to WHO (class and level of evidence 1B) [12].
After 8 weeks the patient did not report any adverse events. 6-minute walk test - increase in distance by 89 m. EchoCG - no dynamics, average pressure in the pulmonary artery - 80 mm Hg. Art. All attempts to increase the dose of Bozenex were accompanied by intense headaches; the patient continued taking the drug at a dose of 62.5 mg 2 times a day.
After 12 months the patient did not report any adverse events. 6-minute walk test - increase in distance compared to the initial visit by 97 m. EchoCG - no dynamics, average pressure in the pulmonary artery - 83 mm Hg. Art. The rest of the therapy remains unchanged.
Specific therapy for PH
The decision to prescribe specific therapy should be made only after catheterization of the right heart with a test for vasoreactivity. If the test result is positive (about 10% of patients with idiopathic PAH), calcium channel blockers are used in high doses. However, only 50% of patients have a stable hemodynamic response to this group of drugs. Features of the prescription and dose of calcium channel blockers for the treatment of PH are presented in Table 4. It should be remembered that the use of this group of drugs empirically, without performing a vasoreactive test, can worsen the clinical condition and prognosis of the patient [10]. Prescribing standard doses of calcium channel blockers is possible only if there are other indications (for example, Raynaud's syndrome, etc.) [1].
In case of a negative test for vasoreactivity, the patient is shown other groups of drugs:
endothelin receptor antagonists (bosentan, ambrisentan, macitentan);
prostanoids (iloprost, epoprostenol (not registered in the Russian Federation));
phosphodiesterase-5 inhibitors (sildenafil, tadalafil, vardenafil);
stimulators of soluble guanylate cyclase (riociguat);
selective prostacyclin IP receptor agonists (selexipag);
combination therapy.
They are expensive and can only be prescribed by an expert center for the treatment of patients with PH. According to the treatment algorithm for PAH in patients with moderate and high risk, the use of combination therapy is preferable (Fig. 1, 2). In this case, in case of insufficient effectiveness, a third drug is added to the combination [10].
The selection of therapy for a patient with PH is carried out by an expert center. After discharge, he needs to report to a regional center or office for patients with PH, where the need for specific therapy will be confirmed by a medical commission. The conclusion of the medical commission and medical documents are then sent to the regional Ministry of Health, where the issue of issuing the necessary medications to the patient is decided.
It should be noted that control of specific therapy is carried out not only by the expert center, but also by regional centers. All complex issues of patient management in the region can be resolved using telemedicine technologies.
Taking into account the high cost of PH therapy, the emergence of high-quality generics can significantly reduce the cost of patient treatment while achieving comparable effectiveness. In 2021, a new drug from the class of endothelin receptor antagonists (bosentan) was registered in the Russian Federation - Bosenex, which has proven bioequivalence with the original drug. However, bioequivalence is not reliable evidence of the effectiveness of a drug.
From December 1, 2017 to September 31, 2018, the Federal State Budgetary Institution National Medical Research Center of Cardiology of the Russian Ministry of Health conducted a study of the safety and effectiveness of Bozenex therapy in patients with PAH for 24 weeks. [eleven]. The study included 42 patients who were initiated on bosentan therapy; 63.6% had not previously received PAH therapy; in 36.4%, the drug was added to therapy with sildenafil at a dose of 60 mg/day. By the 24th week. there was a decrease in the proportion of patients with 3 FC from 55% to 30%, an increase in 2 FC from 45% to 55%, and the appearance of patients with 1 FC (15%); the distance in the 6-minute walk test increased by 52.1 m. During catheterization of the right heart, a positive change in the average pressure in the pulmonary artery (-6.7 mm Hg), average pressure in the right atrium (-1.6 mmHg with achievement of normal values) and pulmonary vascular resistance (-293.2 dyne × s × cm-5) (p <0.05). Bosenex therapy was well tolerated and was not accompanied by clinically significant adverse events [11].
Another proof of the good clinical effectiveness of the drug is a clinical case.