Symptoms
Signs of the disease are different, they depend on where exactly the amyloid deposits are localized, how widespread the disease is, and whether there are complications. Often there is a complex of symptoms reflecting damage to several organs.
With amyloidosis of the gastrointestinal tract the following are observed:
- tongue enlargement;
- difficulty swallowing;
- bowel dysfunction;
- heartburn, nausea;
- stomach ache.
Signs of liver amyloidosis:
- change in liver size;
- pain in the hypochondrium on the right;
- nausea, belching;
- jaundice.
Pancreatic amyloidosis is characterized by dull pain in the left hypochondrium.
Cardiac amyloidosis is expressed in rhythm disturbances, myocardial lesions, and heart failure.
Amyloidosis of the nervous system has the following symptoms:
- peripheral polyneuropathy (numbness of the limbs, tingling, burning sensation);
- headaches, dizziness, increased sweating;
- urinary and fecal incontinence;
- sexual dysfunction.
With amyloidosis of the respiratory system, hoarseness of the voice and bronchitis are observed.
So, what is primary amyloidosis or AL amyloidosis?
Definition of disease.
Amyloid light chain amyloidosis (AL), primary systemic amyloidosis (PSA), or simply primary amyloidosis. The disease occurs when a person's antibody-producing cells (plasmocytes) do not function properly and produce abnormal protein fibers from antibody components called light chains. These light chains form amyloid deposits, which can cause serious damage to various organs. Abnormal light chains in urine are sometimes called "Bence Jones proteins."
Epidemiology.
AL amyloidosis is the most common type of systemic amyloidosis in developed countries, with an estimated incidence of 9 cases per million inhabitants per year. The average age of diagnosed patients is 65 years and less than 10% of patients are under 50 years of age.
Signs and symptoms.
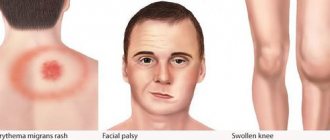
AL amyloidosis can affect a wide range of organs and therefore presents with a range of symptoms. The kidneys are the most commonly affected organ in AL amyloidosis. Symptoms of kidney disease and kidney failure may include fluid retention, swelling, and shortness of breath. Heart complications, which affect more than a third of patients, include heart failure and irregular heartbeat. Other symptoms may include gastrointestinal disturbances, liver enlargement, suppression of adrenal and other endocrine glands, changes in skin color, fatigue and weight loss.
Systemic AL amyloidosis. A. Macroglossia with lateral tongue ridge. B. Bilateral periorbital purpura. C. Pseudo-athletic appearance of secondary diffuse muscle infiltrates. D. Voluminous hepatomegaly due to primary hepatic amyloidosis. E. Diffuse bilateral interstitial lung diseases. F. Enlargement of the submandibular gland. Localized AL amyloidosis. G. Nodular conjunctival amyloidosis. H. Horate amyloid lump.
Diagnosis of the disease.
Diagnosis is based on examination of the area involved showing Congo red-positive amyloid deposits, which stain positive with anti-LC antibody by immunohistochemistry and/or immunofluorescence. Due to the systemic nature of the disease, noninvasive biopsies such as abdominal fat aspiration should be considered before taking biopsies from involved organs to reduce the risk of bleeding.
In addition, blood and urine can be tested for the presence of “light chains,” which can form amyloid deposits, causing disease.
Treatment.
The most effective treatment is autologous bone marrow transplantation using stem cells. However, not all patients are prescribed this type of treatment.
Other treatments may include chemotherapy similar to that used for multiple myeloma. The combination of melphalan and dexamethasone has been found effective in those not suitable for stem cell transplantation, and the combination of bortezomib and dexamethasone is now widespread in clinical use.
Forecast.
Life expectancy with AL amyloidosis depends on the spectrum of organ damage (the main prognostic factor is amyloid heart disease), the severity of damage to individual organs and the hematological response to treatment.
Treatment and prevention
In most cases, amyloidosis is treated at home. If there are complications, the patient may be indicated for hospitalization.
Treatment for amyloidosis includes taking medications and following a number of doctor’s recommendations. But in severe cases, the spleen is removed and kidney or liver transplantation may be required.
The list of medications depends on the location of the deposits, the degree of damage to the body, and existing complications. Thus, with secondary amyloidosis, specific treatment of the primary disease is necessary. In addition, medications are prescribed to eliminate symptoms.
Also, the patient is often prescribed a special diet (restriction of protein and salt intake).
There is no specific preventive program for amyloidosis, since the exact causes of the disease are unknown.
Amyloidosis is a complex disease that requires ongoing treatment. A patient with amyloidosis should be regularly monitored by a specialist and undergo examinations to monitor his health. Medical has modern diagnostic equipment, which allows you to make an accurate diagnosis in the shortest possible time. And the center’s experienced, highly qualified specialists will prescribe effective treatment and ensure proper care for the patient.
RENAL AMYLOIDOSIS
It has been shown that aspiration biopsy of subcutaneous fat allows diagnosing secondary or hereditary amyloidosis in 50-80% of cases, rectal biopsy - in 50-75%. It is especially important that a biopsy of these organs is most often informative already in the proteinuric stage. Therefore, a rectal biopsy should be performed in all unclear cases of proteinuria, as well as in cases of unclear bowel disease, especially those with unexplained diarrhea and/or melena
The problem of amyloidosis has been studied for more than 100 years. The mystery of this disease, in which any organs and tissues can be affected and, consequently, the occurrence of various clinical symptoms, remains not fully solved to this day. The disease got its name due to the fact that the pathological substance formed in the organs resembles starch (due to its ability to be stained with iodine). It is now known that with amyloidosis, a special substance is deposited in the organs - amyloid - an abnormal eosinophilic protein substance that is deposited between the cells of various tissues of the body.
Amyloid in tissues appears either around collagen fibers (pericollagenous amyloidosis) or on basement membranes or around reticular fibers (perireticular amyloidosis).
Table 1. Clinical classification of amyloidosis
1. Primary amyloidosis
2. Secondary amyloidosis
3. Familial (hereditary) amyloidosis
4. Senile amyloidosis 5. Local amyloidosis |
There is no uniform classification of amyloidosis.
The clinical classification is based on its origin (Table 1). Currently, it is customary to distinguish between five main groups of amyloidosis: idiopathic (primary), hereditary (genetic), acquired (secondary, reactive), senile and local. The first four groups of amyloidosis are systemic diseases with predominant damage to any organ. The chemical composition of amyloid and its antigenic properties in different clinical forms of amyloidosis are not the same (Table 2). There are two main chemical classes of amyloid: amyloid consisting of immunoglobulin light chains (AL-type amyloidosis) and amyloid from non-immunoglobulin amyloids (AA-type amyloidosis).
Table 2. Types of amyloid and corresponding forms of amyloidosis
| Types of amyloid | Designation | Precursor protein | Type of amyloidosis |
| Ig light chain amyloid | AL | Monoclonal light chains (kappa or lambda) | 1) Idiopathic generalized 2) With myeloma 3) Types of local |
| Amyloid "A" | AA | Whey protein AAS | 1) Secondary 2) Periodic disease 3) Variants of idiopathic |
| Amyloid in familial amyloidosis | AFP | Homologous prealbumin | Portuguese and other family types |
| Amyloid of endocrine origin | AE AEL AEP | Calcitonin insulin glucagon | Tumors of the APUD system that secrete hormones or pseudohormones |
| Amyloid in senile amyloidosis | AS ASc ASb | Unknown | Senile amyloidosis, senile dementia, Alzheimer's disease |
| Amyloid in patients on dialysis | AN | b2-microglobulin | Amyloidosis in patients on long-term dialysis |
| Amyloid K | AK | Keratin | Skin: spots, papules, lichenification |
| Amyloid in local amyloidosis | AL | Unknown | Local amyloidosis of the skin |
Primary amyloidosis is a disease resulting from dyscrasia of plasma cells, leading to loss of immunoglobulin light chains in organ tissues. In 90% of those suffering from type AL amyloidosis, serum immunoelectrophoresis reveals a monoclonal protein. However, the clone of altered plasma cells in this disease is significantly smaller in volume and activity than in myeloma.
AA amyloid (amyloid “A”) has antigenic similarity to serum protein (a1-globulin), which appears in the serum of patients with inflammatory diseases. This is due to the fact that in the acute phase of inflammation, macrophages produce interleukin-1, which leads to the synthesis of acute phase proteins in the liver, one of which, serum protein SAA, is a precursor of amyloid A.
Primary, or idiopathic, generalized amyloidosis, first described by Wild in 1886, is a generalized process with a predominant lesion of a particular organ or system. The following clinical variants are distinguished: systemic, cardiopathic, neuropathic, nephropathic, etc. The dominant pathology may change throughout the course of the disease.
Secondary amyloidosis occurs against the background of chronic long-term inflammatory diseases (such as rheumatoid arthritis, tuberculosis, osteomyelitis, ulcerative colitis, lymphogranulomatosis, certain tumors, etc.). In most cases, the clinical picture is associated with kidney damage and patients die from chronic renal failure (CRF). In addition, the adrenal glands, liver, spleen and gastrointestinal tract are affected. In accordance with this, some authors distinguish nephropathic, epinephropathic, hepatic and mixed variants of secondary amyloidosis.
Hereditary (genetic, familial) amyloidosis is characterized by the predisposition of ethnic groups to this disease, the special geographic distribution of forms of this amyloidosis, and the presence of this disease in relatives of the same family.
Periodic disease (familial Mediterranean fever) is a hereditary nephropathic form of amyloidosis, transmitted autosomal recessively (chromosome 16) through several generations of families. It manifests itself predominantly in representatives of the so-called ancient nations - primarily in Armenians, Arabs, and Sephardic Jews, although in approximately 50% of cases there may be no family history. Clinical manifestations consist of recurrent serous or fibrinous inflammation of the visceral membranes.
All forms have:
- onset predominantly in early childhood;
- chronic course with exacerbations and remissions;
- stereotypicality of attacks and their benign quality (after attacks of pain
- changes in organs disappear);
- uniformity of laboratory parameters;
- failure of treatment;
- development of amyloidosis with predominant kidney damage;
- characteristic dynamics of kidney damage with a sequential change of stages (preclinical, proteinuic, nephrotic, uremic).
The disease occurs with attacks of pain in the abdomen, chest, etc. Depending on the prevalence of symptoms, four types of disease are distinguished: abdominal, thoracic, articular, febrile. Patients are mistakenly operated on for appendicitis, cholecystitis, pancreatitis, etc. Histological examination of tissues (after operations and biopsies) reveals dilated capillaries with proliferation of the adventitia, leuko-lymphocytic infiltration of the stroma, and effusion with a high fibrin content. During repeated operations, adhesions are found.
The disease before the development of renal amyloidosis has little effect on the general condition of patients. Many of them know that they have “Armenian disease” and show postoperative scars. It is assumed that the basis of the disease is a violation of the metabolism of catecholamines - in patients with familial Mediterranean fever, the level of dopamine-b-hydroxylase is increased outside and during an attack, and in the peritoneal fluid they have a reduced concentration of the complement C5a fragment inhibitor, which is a factor of neutrophil chemotaxis, which may also play a role in causing inflammation during an episode of periodic illness.
Domestic researchers believe that amyloidosis in periodic disease, although represented by type A amyloid, is not a complication, but a manifestation of the same genetic abnormality as the disease itself. This is confirmed by the presence of two independent phenotypes of the disease: in cases belonging to the first phenotype, the disease begins with attacks of periodic disease, and then amyloidosis begins; in the second phenotype, the disease begins with amyloidosis, then attacks of aseptic peritonitis, pleurisy, fever, etc. are added.
At the same time, there are modern studies showing that with a certain haplotype of the familial Mediterranean fever gene, it is possible for it to occur without the development of amyloidosis. Amyloidosis in periodic disease is generalized, but its clinical picture is mainly associated with kidney damage.
Other forms of hereditary amyloidosis, which have an ethnic origin, are much less common in Russia. This variant of hereditary amyloidosis is familial amyloidosis, shown by McClem and Wells using the example of a family dynasty of English industrialists, but, apparently, it can also occur in families of other nationalities. All 9 patients from four generations of the family had a disease that began in early childhood with periodically recurring attacks of fever. In addition to fever, during the attack, patients developed an urticarial rash in the form of itchy, painful reddish papules and Quincke's edema. Over time, the attacks became more frequent and their duration increased. By the age of 20, members of this family developed progressive hearing loss to complete deafness. Later, amyloidosis of the kidneys with all its manifestations developed, as well as amyloidosis of the spleen, liver, adrenal glands and other organs.
Table 3. Hereditary amyloidosis
1. Neuropathic
2. Nephropathic
3. Cardiomyopathic
4. Mixed
|
Neuropathic types of familial amyloidosis have been described in families of Portuguese origin (Andrada type), American, Finnish, etc. These types of amyloidosis result in damage to the kidneys and nerve trunks, which leads to the death of the patient.
These and other forms of hereditary amyloidosis are given in Table. 3. Genetic amyloidosis also occurs in Russians. Thus, in 1969, O. M. Vinogradova and co-authors described hereditary amyloidosis, observed in a Russian family in three generations, which in its clinical course resembled that described by Mackle and Wells.
Senile amyloidosis is detected in older people. It has been shown that it is found in more than 30% of people over 60-70 years of age, in more than 40% of people over 70-80 years of age, and in approximately 80% of people over 80-90 years of age. Amyloidosis can be generalized and local. In this regard, senile systemic (mainly cardiovascular) amyloidosis, senile local (isolated) atrial, cerebral, aortic, pancreatic islet, prostate, seminal vesicle amyloidosis and senile multiorgan amyloidosis are distinguished. Deposition of amyloid in the vessels of the heart, islets of Langerhans and brain amyloidosis (the so-called Schwartz triad) in many cases cause senile degradation, although often senile amyloidosis is not clinically manifested, and amyloid deposits in these organs are found only at autopsy.
Local amyloidosis is a heterogeneous group in terms of amyloid composition. In the form of nodular formations, it can appear in the lungs, larynx, skin, bladder, tongue, etc. Isolated deposits of amyloid are sometimes found in tumors of the APUD system and thyroid gland. Such local forms of amyloidosis can determine the clinical picture of the disease, for example, when localized in the bronchial tree or intestines.
Dialysis amyloidosis (dialysis-associated amyloidosis) has now become a complication of chronic renal failure in patients on long-term hemo- or peritoneal dialysis. The main component of fibrils in this form of amyloidosis is b2-microglobulin. Amyloid is deposited in the ligamentous apparatus of the carpal tunnel and in the bones, causing destructive arthropathy with the formation of subchondral erosions and cysts. Treatment of dialysis amyloidosis consists of surgical removal of the affected tissue and symptomatic therapy. Attempts are being made to create new dialysis membranes that do not allow b2-microglobulin to pass through.
Clinically significant kidney damage is observed mainly in secondary (reactive) amyloidosis and in hereditary forms of amyloidosis, primarily amyloidosis that occurs during periodic illness. In both cases, amyloidosis is of the AA type. In primary generalized amyloidosis, although there is loss of amyloid in the renal tissue, patients die from heart failure or from other causes when nephrotic syndrome or chronic renal failure does not have time to develop. However, the appearance of increasing proteinuria with unclear circulatory failure occurring with cardiomegaly may suggest a diagnosis of primary amyloidosis, and in many cases Bence Jones protein is detected in the urine. In some cases of primary amyloidosis, nephrotic syndrome quickly develops.
Nephrotic syndrome is a characteristic manifestation of renal amyloidosis in familial Mediterranean fever. If nephrotic syndrome develops against the background of a long-term chronic inflammatory disease, such as rheumatoid arthritis, then this is a reliable sign of the addition of reactive amyloidosis with kidney damage.
In fact, edema is preceded by a fairly long preclinical period. Therefore, during the course of renal amyloidosis, several stages are distinguished, which are called differently by different authors.
1. Preclinical (latent, asymptomatic) stage, in which amyloid is present in the intermediary zone and edema and foci of sclerosis develop along the straight vessels of the pyramids. The stage lasts 3-5 or more years. During this period, with reactive amyloidosis, the clinical manifestations of the underlying disease (for example, purulent process in the lungs, tuberculosis, rheumatoid arthritis, etc.) predominate.
2. Proteinuric (albuminuric) stage - amyloid appears primarily in the mesangium, in capillary loops, in the pyramids and cortex of the glomeruli, in the vessels. Sclerosis and nephron atrophy, hyperemia and lymphostasis develop. The buds are enlarged and dense, matte gray-pink in color. Proteinuria at the beginning is moderately expressed, it may even be transient for some period, decrease and increase, but then it becomes persistent (stage of intermittent proteinuria). Some researchers distinguish two periods at this stage: selective and non-selective proteinuria. The duration of the stage is from 10 to 13 years.
3. Nephrotic (edematous, edematous-hypotonic) stage - amyloid-lipoid nephrosis - amyloid in all parts of the nephron. There is sclerosis and amyloidosis of the medulla, but the cortical layer is without pronounced sclerotic changes. Duration of the stage is up to 6 years. In both the proteinuric and nephrotic stages, the kidneys are enlarged and dense (large sebaceous kidney). Clinically, this stage is manifested by classic nephrotic syndrome with all its signs: with the development of massive proteinuria (with loss of protein in the urine of more than 3-5 grams per day), hypoproteinemia with hypoalbuminemia, hypercholesterolemia, lipiduria with edema to the degree of anasarca. In the urinary sediment, hyaline casts are found, and as proteinuria increases, granular casts are found. Possible micro- and macrohematuria, leukocyturia without signs of pyelonephritis.
Nephrotic syndrome with amyloidosis differs from that with glomerulonephritis in that it occurs after a period of prolonged proteinuria, which may not be detected by a doctor, which reduces the value of this symptom. In contrast to glomerulonephritis, amyloidosis is more characterized by an edematous syndrome that occurs with normal or low (in the case of adrenal amyloid infiltration) blood pressure. However, it is now known that nephrotic syndrome and amyloidosis can occur with arterial hypertension.
One of the clinical differential diagnostic signs of amyloid lesions in nephrotic syndrome is the systemic nature of the lesion—identification, along with proteinuria and anasarca, of enlarged lymph nodes, liver and spleen, as well as signs of intestinal damage.
In terms of the frequency of damage in secondary (reactive) amyloidosis, the liver ranks third after the spleen and kidneys and is affected in more than 90% of cases, in hereditary amyloidosis - in 50%. Amyloid liver damage is not characterized by changes in biochemical parameters, except for an increase in alkaline phosphatase, sometimes significant. The spleen is usually enlarged due to amyloid deposition and portal hypertension. Such a combination - nephrotic syndrome and significant enlargement of the liver and spleen with a moderate isolated increase in alkaline phosphatase, signs of damage to the gastrointestinal tract - should suggest inclusion of amyloidosis in the diagnostic algorithm.
4. Uremic (terminal, azotemic) stage - amyloid wrinkled kidney - reduced in size, dense, with scars. Chronic renal failure differs little from that of other kidney diseases. It is believed that, unlike glomerulonephritis, in which the onset of chronic renal failure, occurring with polyuria, can lead to at least partial convergence of edema, with amyloidosis azotemia develops against the background of low blood pressure and nephrotic syndrome.
The clinical picture suggests amyloidosis. However, intravital diagnosis of amyloidosis is based on obtaining via biopsy and examination of histological material of various organs and tissues stained with Congo red or thioflavin.
More invasive are aspiration biopsy of the liver, which allows diagnosing amyloidosis, according to various authors, from 50 to 95%, and kidney, which is informative in 85% of cases. In primary generalized amyloidosis, in which re-collagen loss of amyloid occurs, a biopsy of the gums or tongue may be more informative.
The preparations are stained with Congo red and/or thioflavin T or S. To type AA- and AL-amyloidosis, histological sections of organs are incubated in a solution of potassium permanganate. As a result, the AA protein loses its affinity for Congo red, while the AL protein does not. In addition, AL amyloid is denatured after formalin fixation, whereas AA protein is not denatured and is therefore detected by the immunoperoxidase method.
Treatment of generalized forms of amyloidosis that occurs with kidney damage, primarily secondary and amyloidosis due to periodic illness, most often begins at the stage of nephrotic syndrome. This is due to the fact that their diagnosis is almost always made already during the period of severe edema or even with incipient renal failure. Despite this, one of the central tasks in secondary amyloidosis is the impact on the underlying disease - that is, active therapeutic or surgical treatment of purulent foci or infectious processes. In the diet, in addition to limiting the intake of sodium chloride (table salt), it is suggested to limit foods containing casein. For the symptomatic treatment of nephrotic syndrome and chronic renal failure, as well as for the treatment of these syndromes resulting from kidney damage of other origins, antihypertensive, diuretic drugs, correction of anemia, etc. are indicated.
Specific treatment for AA amyloidosis includes the use of colchicine. The mechanism of action of this drug is still not reliably known; it is assumed that it inhibits the synthesis of SAA protein by hepatocytes. In case of periodic illness, as already mentioned, colchicine is able to prevent its attacks. In case of renal amyloidosis, it reduces proteinuria and, with timely treatment, can completely cure nephrotic syndrome. The drug can cause nausea, vomiting, diarrhea (compensation with enzyme preparations is possible), hair loss (in this case calcium supplements are prescribed), leukopenia, thrombocytopenia, and skin rashes. Therefore, colchicine is prescribed in gradually increasing dosages (up to 2 mg/day), focusing on individual tolerance. Colchicine is taken after successful treatment and after the renal symptoms of amyloidosis have decreased or disappeared, possibly for the rest of the patient's life.
Another drug traditionally used for amyloid nephropathy is unithiol (sodium 2,3-dimercaptopropanesulfonate). The drug binds sulfhydryl groups of proteins - amyloid precursors and thus prevents the formation of amyloid fibrils. Unithiol is able to slow down and stabilize the course of secondary renal amyloidosis. Side effects when using it include nausea, dizziness and tachycardia, so the dose is increased gradually.
For the treatment of secondary amyloidosis that occurs against the background of rheumatoid arthritis, the anti-inflammatory drug dimethyl sulfoxide (Dimexide), which has been used for a long time as an external agent, is also used. For renal amyloidosis, it is prescribed in very small dosages (1-5 mg) orally. The drug is available in bottles in pure form and is diluted before use. Due to the possibility of side effects of dimexide (nausea, vomiting, allergic rashes), the concentration is increased gradually, based on patient tolerance. Because of its unpleasant odor, it is stirred in peach or other fruit juice before use. It is believed that the effectiveness of dimethyl sulfoxide is associated not with its effect on amyloid, but with its anti-inflammatory effect on the primary disease (primarily rheumatoid arthritis).
The basis for the use of 4-aminoquinoline drugs for the treatment of amyloidosis is their inhibitory effect on the synthesis of nucleic acids, inhibition of the formation of acidic mucopolysaccharides, the ability to stabilize cellular and lysosomal membranes and suppress the activity of certain enzymes. From this group of drugs, delagil and plaquenil are mainly used. Both medications are used long-term. As with the prescription of other drugs, side effects are possible when using the 4-aminoquinoline series (nausea, diarrhea, skin changes, psychosis, neutropenia, and in a later period - clouding of the cornea with blurred vision). Discontinuation of the drug, as a rule, leads to regression of these phenomena. It makes sense to use drugs in this series only in the early stages of amyloidosis.
Long-term therapy with raw fried liver does not have a clear pathogenetic basis, but its positive effect on the course of secondary amyloidosis is known. Apply from 80 to 120 mg of liver daily for 6 months - 1 year. One of the complications of this type of treatment is blood eosinophilia, so constant monitoring of the leukocyte count should be carried out.
The basic principles of treatment for AA amyloidosis of the kidney are summarized in Table. 4.
Table 4. Treatment of nephrotic syndrome in secondary amyloidosis and amyloidosis in periodic disease
1. Treatment of the underlying disease 2. Symptomatic treatment of nephrotic syndrome and/or chronic renal failure (limiting the intake of table salt, prescribing diuretics and antihypertensive drugs, transfusion of red blood cells for anemia, correction of electrolyte disturbances, etc.) 3. Diet: protein - 60-70 g/day. (1 g / 1 kg body weight)
4. Colchicine - start with 1 mg/day, increase to 2 mg/day, focusing on tolerability 5. Unithiol - start with 3-5 ml of a 5% solution, gradually increase to 10 ml/day. Course - 30-40 days, repeat the course 2-3 times a year 6. Dimethyl sulfoxide - 1-5% solution of 30-100 ml in fruit juice 7. Delagil - 0.25 mg 2 times a day for 1-2 years 8. Raw fried liver - 80-120 g/day. |
Reactive amyloidosis and amyloidosis during periodic illness often cause the death of a patient from chronic renal failure. But the course of the underlying disease largely determines the prognosis of reactive amyloidosis - frequent exacerbation of the inflammatory or purulent process, the use of corticosteroids sharply accelerate the onset of chronic renal failure. It should be said that the death of a patient with secondary amyloidosis is not always caused by chronic renal failure. Patients with reactive amyloidosis due to rheumatoid arthritis who died due to intestinal damage from diarrhea with malabsorption syndrome are described.
There are great difficulties in treating patients with idiopathic generalized amyloidosis, even with timely diagnosis. The prognosis for these patients is poor—the median survival of those with primary systemic amyloidosis is less than 18 months from diagnosis. Primary systemic amyloidosis is treated with melphalan (0.25 mg/kg body weight per day). It has now been shown that the survival rate of patients with primary systemic amyloidosis is 51% by the end of the first year, 16% in the fifth year, and 4.7% in the tenth year, while the vast majority of patients who lived ten years or more were treated with alkylating agents. Currently, attempts are also being made to prolong the life of these patients through bone marrow transplantation.
Amyloid: morphological characteristics
Amyloid is a glycoprotein in which globular and fibrillar proteins are held together by polysaccharides. Back in 1955, Letterer showed that amyloid is a mixture of two proteins, one of which resembles serum globulins and the other collagen. The severity and irreversibility of amyloidosis is explained by the exceptional strength of the bonds of the protein-polysaccharide components of amyloid with each other and with those tissue elements into which it falls.
Morphologically, amyloid consists of non-branching, ribbon-shaped fibrils (7.5 nm in diameter and 800 nm in length) and periodic rods. The fibrils consist of two subfibrils with a diameter of 2.5 nm. The amyloid substance is built from parallel fibrils consisting of protein and neutral polysaccharides, intertwined with perpendicular fibrils of acidic mucopolysaccharides. Periodic rods make up 5% of the total mass of fibrils. They have a diameter of 10 nm and a length of 250 nm, consist of pentagonal formations and differ in composition from fibrils. Periodic rods are close in their properties to plasma a-globulins.
Amyloid is characterized by an affinity for Congo red and fluorescent dyes such as thioflavin. When stained with Congo red, amyloid is birefringent in polarized light and acquires a green glow on sections (positive anisotropy and dichroism - birefringence). The anisotropy of amyloid is a consequence of its ordered structure. After staining with thioflavin T or S, the amyloid acquires the property of fluorescing in ultraviolet light
Diagnosis of amyloid kidney dystrophy
During the latent stage, diagnosis of the disease is seriously difficult, so laboratory diagnostics of urine and blood are performed. The first make it possible to detect protein and leukocytes, the level of which is constantly increasing, as well as an admixture of blood and bodies from coagulated protein, blood cells, and kidney epithelium. Blood tests detect decreased albumin levels, increased globulin levels, abnormally elevated lipid and/or lipoprotein levels, and electrolyte imbalances.
In the presence of pathological processes, various diagnostic techniques make it possible to identify their specific manifestations. So:
- the electrocardiogram registers arrhythmia;
- echocardiography reveals myocardial pathologies, characterized by structural changes in the heart muscle;
- ultrasound diagnostics of the peritoneum determines an increase in the volume of internal organs such as the liver and spleen;
- X-ray examination of the gastrointestinal tract determines the deterioration of contraction of the stomach walls, too slow or, on the contrary, rapid movement of barium through the intestines;
- Ultrasound scanning of the kidneys reveals their significant increase;
A biopsy of the kidney, rectal mucosa and liver is required.