- Incidence of Ewing's sarcoma
- Risk factors for developing Ewing's sarcoma
- Diagnosis of Ewing's sarcoma
- Determination of the stage (prevalence) of Ewing's sarcoma
- Treatment of Ewing's sarcoma
- After completion of treatment
Ewing's sarcoma ranks second in frequency among malignant bone tumors in children (after osteogenic sarcoma) and was first described by D. Ewing in 1921 and named after the author.
This tumor is rare in children under 5 years of age and in adults over 30 years of age. Most often, this tumor occurs in adolescents aged 10 to 15 years. There are cases of extraosseous Ewing's sarcoma with soft tissue involvement.
Another childhood tumor, primitive neuroectodermal tumor (PNET), shares many features with osseous and extraosseous Ewing sarcoma, which can primarily affect both bone and soft tissue. All of these tumors develop from the same type of cells.
Bone Ewing sarcoma accounts for 87%, extraosseous Ewing sarcoma 8% and PNET -5%.
Typically, such tumors develop in the central part of the long bones of the upper and lower extremities, but can also be detected in the pelvic bones and in the bones and soft tissues that form the chest.
What's happened
This type of sarcoma is a malignant neoplasm that develops from bone tissue. The incidence rate of this type of sarcoma among other malignant bone diseases is 15 percent.
Very often this disease appears in children and adolescents. Boys are most often affected, girls less often. The location of the lesion is long and flat tubular bones. Sometimes this disease occurs not in the bones, but in the soft tissues, called extraosseous Ewing's sarcoma. There are also peripheral primitive neuroectodermal sarcomas, which belong to the varieties of the disease in question.
Ewing's sarcoma is very aggressive and is characterized by the early appearance of metastases. It is characterized by pain, swelling, local fever and enlarged veins near the tumor. The tumor is most often diagnosed in the later stages of development, and pathological fractures often occur at the site of the disease.
It is treated in a combination: surgery, preoperative and postoperative chemotherapy, and radiation therapy.
List of sources
- Fedenko A. A., Bokhyan A. Yu., Gorbunova V. A., Makhson A. N., Teplyakov V. V. Practical recommendations for the treatment of primary malignant bone tumors (osteosarcoma, Ewing sarcoma). Malignant tumors: Practical recommendations RUSSCO #3s2, 2020 (vol. 10).17
- Soloviev Yu.N., Ewing's sarcoma. Oncology Issues 2002; No. 1 pp. 7–16.
- Neustadt E.L. Tumors and tumor-like bone diseases / E.L. Neustadt, A.B. Markochev. - St. Petersburg: Foliant, 2007. - 344 p.
- Tumor and tumor-like processes in children /E. D. Callous [and others]; edited by E. D. Callous. - Minsk: Asar, 2002. - pp. 197–199.
- Tumors and tumor-like bone diseases / Neishtadt E.L., Markochev A.B. - St. Petersburg: Foliot, 2007. - 342 p.
Localization
Ewing's sarcoma is localized in the bones of the pelvis, hip, vertebrae, ribs, shoulder blades, fibula and tibia, and humerus.
When Ewing's sarcoma occurs in long bones, the diaphysis becomes the focus of its spread, and then spreads to the epiphysis. Often the increase in cancer cells occurs along the bone canal.
Most often, metastases in this type of disease occur in the lungs, bone tissue and bone marrow.
In the final stages, metastasis occurs in the central nervous system, lymph nodes and internal organs. When the disease is diagnosed, a large number of people already have metastasis.
Risk factors for developing Ewing's sarcoma
Currently, several risk factors are known to be associated with the occurrence of Ewing's sarcoma.
Floor. Ewing's sarcoma is slightly more common among boys than girls.
Age. In 64% of cases, Ewing's sarcoma occurs between the ages of 10 and 20 years.
Race. Ewing's sarcoma is most often observed in the white population.
Prevention of Ewing's sarcoma, like most other childhood tumors, is currently impossible, since the lifestyle of children and their parents does not affect the incidence of this tumor.
Symptoms
One of the first symptoms is pain in the affected area. At first it is weak, but does not go away at night, during periods of rest, but, on the contrary, intensifies at rest.
Further, the pain becomes stronger, the patient cannot be active during the day, and cannot sleep normally. There is pain on palpation. Local hyperthermia occurs, and the venous network may be dilated.
A few months after the first symptoms, the cancerous tumor grows rapidly and is easily detected by palpation.
In the last stages of the disease, there is a threat of a pathological fracture.
Loss of appetite, weakness, exhaustion of the body, fever, and anemia also occur.
If Ewing's sarcoma is localized in the bones of the lower extremities, the patient may limp.
If the vertebrae are affected, the functions of the pelvic organs may be impaired.
When Ewing's sarcoma is localized in the thoracic region, coughing up blood and difficulty breathing occur.
Causes
The exact causes of the development of Ewing's sarcoma have not been established. Most oncologists believe that hereditary predisposition plays a significant role. Presumably, a high risk factor for the development of the disease is various skeletal anomalies ( enchondroma / aneurysmal bone cyst , etc.) and disorders of the development of the genitourinary system during fetal development ( hypospadias / reduplication of the renal system ). In almost 40% of cases there is a close connection with a previous injury.
Diagnosis
Typically, this pathology is diagnosed after the results of radiography. The image shows that the contours of the tumor are unclear, the cortical plate is stratified.
If the disease involves the periosteum, then x-rays show needle-shaped and lamellar neoplasms. The x-ray will also show a change in soft tissue size with some homogeneity.
Next, after radiography, an examination is carried out by an oncologist, who prescribes a CT or MRI of malignant soft tissues.
These studies determine the size of the tumor, its extent along the bone marrow canals, and its connection with nerves and vessels in adjacent tissues.
To examine the tissue, a biopsy is done: material is taken near the medullary canal or from the soft tissue of the tumor.
Hip cancer
Advantages of Sofia Cancer Center
- unique doctors who treat even complex cases;
- admission and treatment under the compulsory medical insurance policy (check with the center administrators for information);
- excellent equipment: high-tech treatment is carried out using all methods available today;
- numerous certificates and awards, including JCI;
- one of the best diagnostic equipment in Moscow;
- 30 years of unique experience and impeccable reputation;
- increased comfort: cozy, warm interior, cafe and restaurant on the roof with panoramic views - you don’t feel like you’re in a hospital;
- plenty of parking spaces.
“Sofia” is located in the Central Administrative District (CAO) of Moscow, next to several metro stations: “Novoslobodskaya”, “Tverskaya”, “Chekhovskaya”, “Belorusskaya” and “Mayakovskaya”.
You can make an appointment by calling +7 (495) 775-73-60 or through the online form!
Style block “STYLE”
Treatment
Due to the fact that Ewing's sarcoma has an aggressive nature of spread and early metastasis, it is necessary to act on the entire body in a combined manner.
Chemotherapy
There are preoperative and postoperative. Usually several drugs are combined at once. Then they look at the tumor’s response to chemotherapy.
Radiation therapy
Typically, with such therapy, high doses of radiation are immediately prescribed. The site of the disease and the lungs are irradiated if metastases are found there.
Surgical intervention
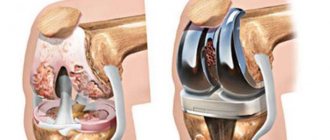
The tumor can be removed entirely along with adjacent soft tissues. If part of the bone is removed, it is replaced with an endoprosthesis. If the tumor cannot be completely removed, it is partially removed.
In modern medicine, they try not to perform disarticulation and amputation, if possible, but to preserve an organ or limb.
Classification by disease stages
The extent of the prevalence of the primary tumor formed the basis for the classification into 4 stages:
- The malignant process does not extend beyond the bone and is well differentiated. Size - up to 8 cm, lymph nodes are not affected.
- The oncological process has not yet spread beyond the bone, is poorly differentiated, and the size exceeds 8 cm.
- In the primary zone there are several foci of bone lesions. Highly malignant degree of differentiation (ability to spread).
- The process has gone beyond the bone.
Prevention of bone cancer
There are no specific methods for preventing bone neoplasm. As general measures we can recommend:
- Quit smoking and abusing alcoholic beverages.
- Lead a physically active lifestyle.
- Maintain immunity: taking herbal preparations that stimulate the immune system, hardening water procedures, regularly taking vitamin-mineral complex tablets.
- Stick to a balanced diet.
- Regularly undergo preventive medical examinations.
However, it should be borne in mind that a healthy lifestyle does not guarantee a reduction in the risk of developing any types of malignant diseases. Therefore, if you suspect bone cancer, even mild symptoms should alert you. Seeking specialized professional medical help allows you to start therapy early and increase the effectiveness of treatment measures.
Diagnostics
If osteogenic sarcoma is suspected, a diagnostic search is carried out by specialists such as a pediatric oncologist, a pediatric surgeon, a pathologist, and a radiologist. The final diagnosis is established based on the results of histological examination.
At the first meeting with a young patient, the doctor collects an anamnesis, clarifies information about the presence or absence of any chronic diseases of the musculoskeletal system, previous injuries, surgical interventions, hereditary syndromes, and family hereditary oncological pathology. A clinical examination of the patient includes an assessment of the focus of the affected limb, regional lymph nodes, and physical status (general condition of the patient).
The diagnostic search consists of the following studies:
- radiography of the affected area;
- computed tomography (CT) with contrast enhancement of the tumor-affected area and chest organs;
- osteoscintigraphy;
- Magnetic resonance imaging of the affected area.
The most common and simplest method for diagnosing osteogenic sarcoma, which can be performed on an outpatient basis, is an x-ray examination of the affected limb. Situations cannot be excluded when the formation is an accidental finding during radiography performed for a traumatic injury. In such situations, the patient is indicated for further examination.
Characteristic radiological signs of osteogenic sarcoma (Fig. 2):
- destruction and sclerosis of bone tissue in the affected area;
- Codman's periosteal visor, which is the periosteum exfoliated by the tumor;
- needle periostitis with multiple bone spicules;
- foci of pathological bone formation in soft tissues (fields of ossification) in the area of formation;
- pathological fracture of the affected limb.
Rice. 2. Radiological signs of osteosarcoma
For clearer visualization of the process, clarification of the size of the formation, spread, and exclusion of soft tissue damage, children are advised to perform computed tomography. In addition, this method allows you to evaluate the primary tumor and detect the presence of distant metastatic foci.
Skeletal scintigraphy has long been the standard method for diagnosing bone lesions, being more sensitive than x-ray methods. It allows you to study the entire human body in a short period of time. This study is based on an increase in the accumulation of phosphorus compounds in the lesion, which indicates the activity of a process characteristic of malignant neoplasms (Fig. 3).
Fig.3. Pathological accumulation of radiopharmaceutical (Technetium mTc)
Magnetic resonance imaging (MRI) of the primary tumor lesion involving the adjacent joint is the optimal method for local staging. MRI monitoring against the background of systemic therapy makes it possible to assess the dynamics of the process and, accordingly, plan the scope of surgical intervention in the future.
One of the key stages of diagnosis is a biopsy of the formation and subsequent histological and immunohistochemical examination of the obtained material.
There are two different approaches to obtaining material: using a trephine needle and an open type of biopsy. The manipulation is performed under CT or ultrasound navigation or using an electron-optical converter (Fig. 4).
Fig. 4: Biopsy using a trephine needle
Chapter 13.
Ewing's sarcoma is the second most common malignant bone tumor in children, accounting for 10-15%. This tumor is rare in children under 5 years of age and in adults over 30 years of age. The peak incidence occurs between 10 and 15 years.
Ewing's sarcoma consists of small round cells with scanty cytoplasm, a round nucleus containing delicate chromatin and faintly visible basophilic nucleoli. Unlike osteosarcoma, it does not produce osteoid.
There is some connection between the occurrence of Ewing's sarcoma and the presence of skeletal abnormalities (enchondroma, aneurysmal bone cyst, etc.) and abnormalities of the genitourinary system (hypospadias, reduplication of the renal system). Unlike osteosarcoma, ionizing radiation is not associated with the occurrence of Ewing's sarcoma.
Cytogenetic analysis shows in 85% of cases the chromosomal translocation t(11,22) (q24,q12) in the majority of cells isolated from this tumor. Similar changes are detected in another small cell tumor, PNET (primitive neuroectodermal tumor). And although Ewing's sarcoma has no anatomical connection with the structures of the central nervous system or the autonomic sympathetic nervous system, these cytogenetic changes prove the neuroectodermal nature of the tumor. In addition, in most cases, expression of the PAX3 protein is detected in tumor cells, which is normally detected during the embryonic development of neuroectodermal tissue. In Ewing's sarcoma, another tumor marker, NSE (neuron-specific serum enolase), can often be detected.
Ewing's sarcoma is one of the most aggressive malignant tumors. Before the use of systemic therapy, almost 90% of patients developed metastases. The most common locations of metastases at the time of initial diagnosis are lungs, bones, and bone marrow. 14%-50% of patients already have metastases detected by routine research methods by the time of diagnosis, and many more patients have micrometastases. Lymphogenous spread of metastases is rare and is always associated with a poor prognosis. Rarely, retroperitoneal and mediastinal spread of metastases also occurs. 2.2% of patients have metastases in the central nervous system during primary diagnosis, and almost all of them have metastases in the generalization of the process.
Clinic
Clinical signs of Ewing's sarcoma are increasing pain, swelling over the affected area with impaired limb function. The tumor is usually painful on palpation and quickly increases in size. Damage to peripheral nerves can cause neurological symptoms. Fever of varying degrees may occur. The soft tissue component of the tumor is often more pronounced than the bone lesion.
Hemorrhages and necrosis are often found in the tumor, which causes an increase in local temperature, erythema and imitates nonspecific inflammation, which complicates diagnosis. Such symptoms suggest, first of all, the presence of osteomyelitis.
The most common location of Ewing's sarcoma is the pelvic bones, femur, tibia, fibula, ribs, scapula, vertebrae, humerus. In contrast to osteogenic sarcoma, Ewing's sarcoma most often affects flat bones (Figure 13-1). In tubular bones, the tumor is localized mainly in the diaphysis and tends to spread to the epiphyses of the bone. In 91% of cases, the tumor is located intramedullary, and the spread along the medullary canal is often greater than through the bone.
X-ray signs of Ewing's sarcoma.
-Destruction of bone (“moth-eaten”) without clear boundaries with a tendency to spread along the medullary canal (Fig. 13-2). - “Bulbous periostitis” is a multilayer linear periostitis, which can be combined with needle periostitis (Fig. 13-3). -In 5% of cases a pathological fracture is detected. When the tumor is localized in the proximal femur, a pathological fracture occurs much more often (in more than 70% of cases). -X-ray examination of soft tissues reveals a distinct soft tissue component of a homogeneous structure. -Damage to the ribs is often combined with pleurisy.
But, nevertheless, radiological signs are not absolutely pathognomonic. It is necessary to carry out differential diagnosis with other pathological processes in the bones - with osteomyelitis (primarily), trauma, other malignant bone tumors (rhabdomyosarcoma, synovial sarcoma, lymphoma, neuroblastoma).
Diagnostics
Carrying out all diagnostic procedures is necessary to verify the diagnosis, determine the extent of the process and identify risk factors, which, in turn, determines the patient’s treatment tactics.
1. Tumor biopsy. A trephine biopsy of the tumor is performed. A sufficient amount of material can sometimes be obtained from the soft tissue component. If this is not possible, the material is obtained from the area of bone adjacent to the medullary canal. It is better to perform a biopsy on the anterior, posterior or medial surface of the limb rather than on the lateral (since the risk of pathological fracture increases). The biopsy site, as well as all wound drainage sites, must subsequently be included in the irradiation zone. 2. X-ray of the lesion. 3. X-ray and CT scan of the lungs 4. Aspiration biopsy or trephine biopsy of the bone marrow from several places (since Ewing’s sarcoma tends to metastasize to the bone marrow) 5. Bone scintigraphy with Te99 - allows to identify other foci in the bones, since there may be multiple bone metastases. 6. Computed tomography and/or nuclear magnetic resonance imaging of the lesion - most accurately determines the size of the tumor, its connection with surrounding tissues, the neurovascular bundle, and the spread of the tumor along the bone marrow canal. 7. Angiography 8. Ultrasound examination
In modern laboratories, cytogenetic and molecular biological studies of the tumor are carried out, and for this, material obtained from aspiration biopsy is often sufficient. Molecular biological studies are also used to identify “minimal residual tumor” (for this, the presence of 1 tumor cell per 100,000 normal cells is sufficient, whereas microscopically tumor infiltration in the bone marrow can only be detected in the presence of 5%-10% of tumor cells).
Unfavorable prognostic factors: -metastases in bone and bone marrow (survival rate for this group of patients is less than 10%); -high level of LDH; - damage to the pelvic bones (especially with high levels of LDH); - metastases to lymph nodes; - the volume of the tumor mass (including the soft tissue component) is 100 ml or more. According to the CESS-86 study, 7-8-year disease-free survival in patients with a tumor volume of less than 100 ml was 63%, and in patients with a tumor volume of 200 ml or more - only 34%.
Unlike osteogenic sarcoma, a pathological fracture does not affect the prognosis of the disease.
Treatment
Before the era of effective systemic therapy, approximately 90% of patients with primary Ewing's sarcoma of bone died within the first 2 to 5 years of disease from systemic pulmonary, bone marrow, and/or bone metastases, despite the fact that, unlike osteosarcoma, this tumor is sensitive to radiation therapy. An impressive improvement in survival rate was achieved with the start of chemotherapy - more than 50% (for Ewing's sarcoma, survival rates are determined at 7-8 years of remission).
Therapy for Ewing's sarcoma includes: 1. Multicomponent chemotherapy (drugs used - vincristine, adriamycin, ifosfamide, cyclophosphamide, actinomycin, Vepesid in combination). Modern treatment programs use preoperative and postoperative polychemotherapy, which also takes into account the histological response of the tumor to treatment. A good tumor response to chemotherapy is considered to be the presence of less than 5% of living tumor cells (EICESS data, EW-93). 2. Radiation therapy to the lesion in high doses - 45-55 Gy (see Chapter Radiation therapy). If metastases develop in the lungs, radiation therapy to the lungs is performed. 3. If possible, radical removal of the tumor (including bone and soft tissue component). Radical resection is possible for lesions in the fibula, forearm bones, ribs, collarbone, and scapula.
Surgery improves local tumor control. When combined with intensive chemotherapy and radiation therapy, complete tumor resection significantly reduces the risk of local recurrence. A decrease in the incidence of local recurrence is observed even after non-radical operations. Modern surgical technology makes it possible to perform organ-preserving operations in cases of damage to the femur and humerus, as well as resection of the pelvic bones.
Patients with a poor prognosis, in particular those with metastases to the bones and bone marrow, with a survival rate of less than 10%, have recently been treated with more intensive treatment - chemotherapy with megadoses of drugs with total body irradiation and transplantation of autologous bone marrow or peripheral stem cells.
This therapy can cure more than 30% of patients with an advanced process (with metastases to the bones and bone marrow). In patients with good tumor sensitivity to chemotherapy, even better treatment results can be achieved (7-year survival rate is about 50%). Page 196